Lenalidomide OHRE
Lenalidomide is used to treat different types of cancers.
Dosage: 5mg, 10mg, 15mg, 25mg
")
")
Cliquez sur un pictogramme pour aller directement à la rubrique le concernant.
Pour plus d'information sur les pictogrammes, consultez l'aide.
Indications thérapeutiques
Classe pharmacothérapeutique : IMMUNOSUPPRESSEURS, AUTRES IMMUNOSUPPRESSEURS - Code ATC : L04AX04.
Qu’est-ce que LENALIDOMIDE STRAGEN 10 mg, gélule
LENALIDOMIDE STRAGEN 10 mg, gélule contient la substance active « lénalidomide ». Ce médicament appartient à un groupe de médicaments qui modifient le fonctionnement de votre système immunitaire.
Dans quels cas LENALIDOMIDE STRAGEN 10 mg, gélule
LENALIDOMIDE STRAGEN 10 mg, gélule est utilisé chez les patients adultes dans le traitement :
· du myélome multiple
· des syndromes myélodysplasiques
· du lymphome à cellules du manteau
· du lymphome folliculaire
Myélome multiple
Le myélome multiple est un type de cancer touchant un certain type de cellules sanguines appelées plasmocytes. Ces cellules s’accumulent dans la moelle osseuse et se multiplient et deviennent incontrôlées. Cela peut entraîner une atteinte des os et des reins.
En général, le myélome multiple ne peut pas être guéri. Cependant, les signes et symptômes peuvent régresser de façon importante ou disparaître pendant une certaine période. Cela est appelé une « rémission ».
Myélome multiple non préalablement traité - chez les patients qui ont reçu une greffe de moelle osseuse
Dans cette indication, LENALIDOMIDE STRAGEN 10 mg, gélule est utilisé Dans cette indication, traitement d’entretien après une récupération adéquate des patients à la suite d’une greffe de moelle osseuse.
Myélome multiple non préalablement traité – chez les patients qui ne peuvent pas être traités par une greffe de moelle osseuse
LENALIDOMIDE STRAGEN 10 mg, gélule est pris avec d’autres médicaments. Ceux-ci peuvent être :
· un médicament de chimiothérapie appelé « bortézomib »
· un médicament anti-inflammatoire appelé « dexaméthasone »
· un médicament de chimiothérapie appelé « melphalan » et
· un médicament anti-inflammatoire appelé « prednisone »
Vous prendrez ces autres médicaments au début du traitement et vous continuerez ensuite en prenant LENALIDOMIDE STRAGEN 10 mg, gélule seul.
Si vous êtes âgé(e) de 75 ans ou plus ou si vous présentez des troubles rénaux modérés à sévères, votre médecin effectuera une évaluation attentive avant le début du traitement.
Myélome multiple - chez les patients qui ont déjà été traités
LENALIDOMIDE STRAGEN 10 mg, gélule est pris en association avec un médicament anti-inflammatoire appelé « dexaméthasone ».
LENALIDOMIDE STRAGEN 10 mg, gélule peut empêcher l’aggravation des signes et symptômes du myélome multiple. Il a également été démontré qu’il retarde la récidive du myélome multiple après le traitement.
Syndromes myélodysplasiques (SMD)
Le terme SMD désigne un ensemble de nombreuses maladies différentes du sang et de la moelle osseuse. Les cellules sanguines deviennent anormales et ne fonctionnent pas correctement. Les patients peuvent présenter différents signes et symptômes, notamment un taux faible de globules rouges (anémie), un besoin de transfusions sanguines et un risque d’infection.
LENALIDOMIDE STRAGEN 10 mg, gélule est utilisé seul pour traiter les patients adultes chez lesquels un SMD a été diagnostiqué et lorsque toutes les conditions ci-dessous sont remplies :
· vous avez besoin de transfusions sanguines régulières pour corriger un taux faible de globules rouges (« anémie avec dépendance transfusionnelle »)
· vous présentez une anomalie des cellules dans la moelle osseuse appelée « anomalie cytogénétique de délétion 5q isolée ». Cela signifie que votre organisme ne fabrique pas assez de cellules sanguines saines
· d’autres traitements qui ont été administrés préalablement, ne sont pas adaptés ou ne sont pas suffisamment efficaces
LENALIDOMIDE STRAGEN 10 mg, gélule peut augmenter le nombre de globules rouges normaux produits par l’organisme en diminuant le nombre de cellules anormales :
· Cela peut réduire le nombre de transfusions sanguines nécessaires. Il est possible que le recours aux transfusions ne soit plus nécessaire.
Lymphome à cellules du manteau (LCM)
Le LCM est un cancer d‘une partie du système immunitaire (le tissu lymphoïde). Il touche un type de globules blancs appelés lymphocytes B ou cellules B. Le LCM est une maladie dans laquelle les lymphocytes B se multiplient de façon incontrôlée et s’accumulent dans le tissu lymphoïde, la moelle osseuse ou le sang.
LENALIDOMIDE STRAGEN 10 mg, gélule est utilisé seul pour traiter les patients adultes qui ont été préalablement traités avec d’autres médicaments.
Lymphome folliculaire (LF)
Le LF est un cancer à progression lente qui touche les lymphocytes B, un type de globules blancs qui aident votre organisme à lutter contre les infections. En cas de LF, un nombre excessif de ces lymphocytes B peut s’accumuler dans le sang, la moelle osseuse, les ganglions lymphatiques et la rate.
LENALIDOMIDE STRAGEN 10 mg, gélule est pris en association avec un autre médicament appelé « rituximab » pour le traitement des patients adultes atteints de lymphome folliculaire préalablement traité.
Comment agit LENALIDOMIDE STRAGEN 10 mg, gélule ?
LENALIDOMIDE STRAGEN 10 mg, gélule agit en modifiant le fonctionnement du système immunitaire de l’organisme et en attaquant directement le cancer. Il agit de plusieurs façons différentes :
· en arrêtant le développement des cellules cancéreuses,
· en arrêtant la croissance des vaisseaux sanguins dans la tumeur,
· en stimulant une partie du système immunitaire pour attaquer les cellules cancéreuses.
Groupe(s) générique(s)
Ce médicament appartient au(x) groupe(s) générique(s) suivants :
Composition en substances actives
-
 Gélule (Composition pour une gélule)
Gélule (Composition pour une gélule)
-
> lénalidomide
 10 mg
10 mg
-
> lénalidomide
Présentations
> plaquette(s) OPA : polyamide orienté aluminium PVC-Aluminium de 21 gélule(s)
Code CIP : 34009 302 049 5 5Déclaration de commercialisation : 24/02/2022
Cette présentation est agréée aux collectivités
Inscription sur la liste de rétrocession au titre de son AMM, selon les conditions précisées au Journal Officiel. Prix de cession publié au Journal Officiel.
Service médical rendu (SMR) 
Ce médicament étant un générique, le SMR n'a pas été évalué par la commission de la transparence (CT), il est possible de se référer à la /aux spécialité(s) de référence du groupe générique auquel appartient ce médicament (cliquez ici pour aller à la rubrique des groupes génériques)
Amélioration du service médical rendu (ASMR)
Ce médicament étant un générique, l'ASMR n'a pas été évalué par la commission de la transparence (CT), il est possible de se référer à la /aux spécialité(s) de référence du groupe générique auquel appartient ce médicament (cliquez ici pour aller à la rubrique des groupes génériques)
Autres informations (cliquer pour afficher)
- Titulaire de l'autorisation : STRAGEN-France
-
Conditions de prescription et de délivrance :
- liste I
- médicament nécessitant une surveillance particulière pendant le traitement
- pour les femmes susceptibles de procréer : surveillance particulière pendant le traitement
- prescription hospitalière
- prescription nécessitant préalablement le recueil de l’accord de soins du patient
- prescription réservée aux médecins compétents en CANCEROLOGIE
- prescription réservée aux médecins compétents en maladie du sang
- prescription réservée aux spécialistes et services HEMATOLOGIE
- prescription réservée aux spécialistes et services ONCOLOGIE MEDICALE
- Statut de l'autorisation : Valide
- Type de procédure : Procédure décentralisée
- Code CIS : 6 620 589 9
ANSM - Mis à jour le : 26/07/2024
LENALIDOMIDE STRAGEN 10 mg, gélule
2. COMPOSITION QUALITATIVE ET QUANTITATIVE 
Chaque gélule contient 10 mg de lénalidomide.
Excipients à effet notoire :
Chaque gélule contient 132,9 mg de lactose.
Pour la liste complète des excipients, voir rubrique 6.1.
Corps jaune opaque et tête verte à vert clair opaque, avec une longueur d’approximativement 21,7 mm, portant l’inscription « L9NL » et « 10 ».
4.1. Indications thérapeutiques
Myélome multiple
LENALIDOMIDE STRAGEN 10 mg, gélule est indiqué en monothérapie pour le traitement d'entretien du myélome multiple non préalablement traité chez les patients adultes ayant reçu une autogreffe de cellules souches.
LENALIDOMIDE STRAGEN 10 mg, gélule est indiqué en association avec la dexaméthasone, ou le bortezomib et la dexaméthasone, ou le melphalan et la prednisone (voir rubrique 4.2), pour le traitement du myélome multiple non préalablement traité chez les patients adultes non éligibles à une greffe.
LENALIDOMIDE STRAGEN 10 mg, gélule est indiqué, en association avec la dexaméthasone, pour le traitement du myélome multiple chez les patients adultes ayant déjà reçu au moins un traitement antérieur.
Syndromes myélodysplasiques
LENALIDOMIDE STRAGEN 10 mg, gélule est indiqué en monothérapie pour le traitement des patients adultes présentant une anémie avec dépendance transfusionnelle due à un syndrome myélodysplasique à risque faible ou intermédiaire 1 associé à une anomalie cytogénétique de type délétion 5q isolée, lorsque les autres options thérapeutiques sont insuffisantes ou inappropriées.
Lymphome à cellules du manteau
LENALIDOMIDE STRAGEN 10 mg, gélule est indiqué en monothérapie pour le traitement des patients adultes présentant un lymphome à cellules du manteau en rechute ou réfractaire (voir rubriques 4.4 et 5.1).
Lymphome folliculaire
LENALIDOMIDE STRAGEN 10 mg, gélule est indiqué, en association avec le rituximab (anticorps anti-CD20), pour le traitement des patients adultes présentant un lymphome folliculaire (Grade 1 – 3a) préalablement traité.
4.2. Posologie et mode d'administration
Dans toutes les indications présentées ci-dessous :
· La posologie est modifiée en fonction des résultats des examens cliniques et des analyses biologiques (voir rubrique 4.4).
· Il est recommandé d’ajuster la posologie pendant le traitement et lors de la reprise du traitement pour prendre en charge les thrombopénies ou neutropénies de Grade 3 ou 4, ou les autres toxicités de Grade 3 ou 4 jugées comme étant liées au lénalidomide.
· En cas de neutropénie, l’utilisation de facteurs de croissance pour la prise en charge des patients devra être envisagée.
· Si moins de 12 heures se sont écoulées depuis la dose manquante, le patient peut prendre la dose. Si plus de 12 heures se sont écoulées depuis la dose manquante à l’heure habituelle, le patient ne doit pas prendre la dose, mais prendre la prochaine dose à l’heure habituelle le lendemain.
Posologie
· Myélome multiple non préalablement traité (MMNPT)
Lénalidomide administré en association avec la dexaméthasone jusqu’à la progression de la maladie chez les patients non éligibles à une greffe
Le traitement par le lénalidomide ne doit pas être initié si la numération des polynucléaires neutrophiles (PNN) est < 1,0 x 109/L et/ou si la numération plaquettaire est < 50 x 109/L.
Posologie recommandée
La dose initiale recommandée est de 25 mg de lénalidomide par voie orale en une prise par jour les jours 1 à 21 de chaque cycle de 28 jours. La dose recommandée de dexaméthasone est de 40 mg en une prise par jour par voie orale les jours 1, 8, 15 et 22 de chaque cycle de 28 jours. Le patient peut poursuivre le traitement par le lénalidomide et la dexaméthasone jusqu’à la progression de la maladie ou une toxicité.
· Paliers de réduction de la posologie
|
|
Lénalidomidea |
Dexaméthasonea |
|
Dose initiale |
25 mg |
40 mg |
|
Palier de dose -1 |
20 mg |
20 mg |
|
Palier de dose -2 |
15 mg |
12 mg |
|
Palier de dose -3 |
10 mg |
8 mg |
|
Palier de dose -4 |
5 mg |
4 mg |
|
Palier de dose -5 |
2,5 mg |
Sans objet |
ª Les réductions de la posologie des deux médicaments peuvent être gérées indépendamment.
· Thrombopénie
|
Numération plaquettaire |
Action recommandée |
|
Chute < 25 x 109/L |
Arrêter l’administration de lénalidomide pendant le reste du cycleª |
|
Retour > 50 x 109/L |
Diminuer d’un palier de dose lors de la reprise du dosage au prochain cycle |
a En cas de toxicité dose-limitante (TDL) survenant à partir du 15e jour d’un cycle, le traitement par le lénalidomide doit être interrompu pendant au moins le reste du cycle de 28 jours en cours.
· Numération des polynucléaires neutrophiles (PNN) - neutropénie
|
Numération des PNN |
Action recommandéea |
|
Première chute < 0,5 x 109/L |
Interrompre le traitement par le lénalidomide |
|
Retour ≥ 1 x 109/L quand la neutropénie est la seule toxicité observée |
Reprendre le lénalidomide à la dose initiale en une prise par jour |
|
Retour ≥ 0,5 x 109/L, quand des toxicités hématologiques dépendantes de la dose autres qu’une neutropénie sont observées |
Reprendre le lénalidomide au palier de dose -1 en une prise par jour |
|
Pour toute nouvelle rechute < 0,5 x 109/L |
Interrompre le traitement par le lénalidomide |
|
Retour ≥ 0,5 x 109/L |
Reprendre le lénalidomide au palier de dose immédiatement inférieur en une prise par jour. |
a A l’appréciation du médecin, si la neutropénie est la seule toxicité quel que soit le palier de dose, ajouter un facteur de croissance granulocytaire (G CSF) et maintenir la dose de lénalidomide.
En cas de toxicité hématologique, la dose de lénalidomide peut être repris au palier de dose immédiatement supérieur (jusqu’à la dose initiale) après amélioration de la fonction médullaire (absence de toxicité hématologique pendant au moins 2 cycles consécutifs : PNN ≥ 1,5 x 109/L et plaquettes ≥ 100 x 109/L au début d’un nouveau cycle).
Lénalidomide en association avec le bortézomib et la dexaméthasone suivis d’un traitement par le lénalidomide et la dexaméthasone jusqu’à la progression de la maladie chez les patients non éligibles à une greffe
Traitement initial : lénalidomide en association avec le bortézomib et la dexaméthasone
Le traitement par le lénalidomide en association avec le bortézomib et la dexaméthasone ne doit pas être initié si la numération des PNN est < 1,0 x 109/L et/ou si la numération plaquettaire est < 50 x 109/L.
La dose initiale recommandée est de 25 mg de lénalidomide par voie orale en une prise par jour les jours 1 à 14 de chaque cycle de 21 jours, en association avec le bortézomib et la dexaméthasone. Le bortézomib doit être administré en injection sous-cutanée (1,3 mg/m2 de surface corporelle) deux fois par semaine les jours 1, 4, 8 et 11 de chaque cycle de 21 jours. Pour des informations supplémentaires sur la dose, le schéma posologique et les ajustements de la posologie des médicaments administrés avec le lénalidomide, voir la rubrique 5.1 et le Résumé des Caractéristiques du Produit de chaque médicament.
Jusqu’à 8 cycles de traitement de 21 jours (durée du traitement initial de 24 semaines) sont recommandés.
Poursuite du traitement : lénalidomide en association avec la dexaméthasone jusqu’à la progression de la maladie
Poursuivre le traitement par le lénalidomide 25 mg par voie orale en une prise par jour les jours 1 à 21 de cycles de 28 jours répétés, en association avec la dexaméthasone. Le traitement doit être poursuivi jusqu’à progression de la maladie ou survenue d’une toxicité inacceptable.
· Paliers de réduction de la posologie
|
|
Lénalidomidea |
|
Dose initiale |
25 mg |
|
Palier de dose -1 |
20 mg |
|
Palier de dose -2 |
15 mg |
|
Palier de dose -3 |
10 mg |
|
Palier de dose -4 |
5 mg |
|
Palier de dose -5 |
2,5 mg |
ª Les réductions de la posologie des deux médicaments peuvent être gérées indépendamment.
· Thrombopénie
|
Numération plaquettaire |
Action recommandée |
|
Chute < 30 x 109/L |
Interrompre le traitement par le lénalidomide |
|
Retour ≥ 50 x 109/L |
Reprendre le lénalidomide au palier de dose -1 en une prise par jour |
|
Pour toute nouvelle rechute < 30 x 109/L |
Interrompre le traitement par le lénalidomide |
|
Retour ≥ 50 x 109/L |
Reprendre le lénalidomide au palier de dose immédiatement inférieur en une prise par jour |
· Numération des polynucléaires neutrophiles (PNN) - neutropénie
|
Numération des PNN |
Action recommandéea |
|
Chute < 0,5 x 109/L |
Interrompre le traitement par le lénalidomide |
|
Retour ≥ 1 x 109/L quand la neutropénie est la seule toxicité observée |
Reprendre le lénalidomide à la dose initiale en une prise par jour |
|
Retour ≥ 0,5 x 109/L, quand des toxicités hématologiques dépendantes de la dose autres qu’une neutropénie sont observées |
Reprendre le lénalidomide au palier de dose -1 en une prise par jour |
|
Pour toute nouvelle rechute < 0,5 x 109/L |
Interrompre le traitement par le lénalidomide |
|
Retour ≥ 0,5 x 109/L |
Reprendre le lénalidomide au palier de dose immédiatement inférieur en une prise par jour. |
a A l’appréciation du médecin, si la neutropénie est la seule toxicité quel que soit le palier de dose, ajouter un facteur de croissance granulocytaire (G CSF) et maintenir la dose de lénalidomide.
Lénalidomide en association avec le melphalan et la prednisone suivis d’un traitement d’entretien par le lénalidomide chez les patients non éligibles à une greffe
Le traitement par le lénalidomide ne doit pas être initié si la numération des PNN est < 1,5 x 109/L et/ou si la numération plaquettaire est < 75 x 109/L.
Posologie recommandée
Les doses initiales recommandées sont de 10 mg de lénalidomide par voie orale en une prise par jour les jours 1 à 21 de chaque cycle de 28 jours pendant 9 cycles au maximum, de 0,18 mg/kg de melphalan par voie orale les jours 1 à 4 de chaque cycle de 28 jours et de 2 mg/kg de prednisone par voie orale les jours 1 à 4 de chaque cycle de 28 jours. Les patients ayant terminé 9 cycles ou qui ne peuvent pas terminer le traitement en association en raison d’une toxicité sont traités par le lénalidomide en monothérapie comme suit : 10 mg par voie orale en une prise par jour les jours 1 à 21 de chaque cycle de 28 jours jusqu’à la progression de la maladie.
· Paliers de réduction de la posologie
|
|
Lénalidomide |
Melphalan |
Prednisone |
|
Dose initiale |
10 mgª |
0,18 mg/kg |
2 mg/kg |
|
Palier de dose -1 |
7,5 mg |
0,14 mg/kg |
1 mg/kg |
|
Palier de dose -2 |
5 mg |
0,10 mg/kg |
0,5 mg/kg |
|
Palier de dose -3 |
2,5 mg |
Sans objet |
0,25 mg/kg |
a Si la neutropénie est la seule toxicité quel que soit le palier de dose, ajouter un facteur de croissance granulocytaire (G-CSF) et maintenir la dose de lénalidomide.
· Thrombopénie
|
Numération plaquettaire |
Action recommandée |
|
Chute < 25 x 109/L |
Interrompre le traitement par le lénalidomide |
|
Retour ≥ 25 x 109/L |
Reprendre le lénalidomide et le melphalan au palier de dose -1 |
|
Pour toute nouvelle rechute < 30 x 109/L |
Interrompre le traitement par le lénalidomide |
|
Retour ≥ 30 x 109/L |
Reprendre le lénalidomide au palier de dose immédiatement inférieur (palier de dose -2 ou -3) en une prise par jour. |
· Numération des polynucléaires neutrophiles (PNN) - neutropénie
|
Numération des PNN |
Action recommandéea |
|
Première chute < 0,5 x 109/L |
Interrompre le traitement par le lénalidomide |
|
Retour ≥ 0,5 x 109/L quand la neutropénie est la seule toxicité observée |
Reprendre le lénalidomide à la dose initiale en une prise par jour |
|
Retour ≥ 0,5 x 109/L, quand des toxicités hématologiques dépendantes de la dose autres qu’une neutropénie sont observées |
Reprendre le lénalidomide au palier de dose -1 en une prise par jour |
|
Pour toute nouvelle rechute < 0,5 x 109/L |
Interrompre le traitement par le lénalidomide |
|
Retour ≥ 0,5 x 109/L |
Reprendre le lénalidomide à la dose immédiatement inférieure en une prise par jour. |
ª A l’appréciation du médecin, si la neutropénie est la seule toxicité quel que soit le palier de dose, ajouter un facteur de croissance granulocytaire (G CSF) et maintenir la dose de lénalidomide.
Lénalidomide en traitement d’entretien chez les patients ayant reçu une autogreffe de cellules souches (AGCS)
Le traitement d’entretien par le lénalidomide doit être instauré après une bonne récupération hématologique post-AGCS, chez les patients sans signe de progression. Le traitement par le lénalidomide ne doit pas être initié si la numération des PNN est < 1,0 x 109/L, et/ou si la numération plaquettaire est < 75 x 109/L.
Posologie recommandée
La dose initiale recommandée est de 10 mg de lénalidomide par voie orale en une prise par jour de façon continue (les jours 1 à 28 de chaque cycle de 28 jours) jusqu’à la progression de la maladie ou survenue d’une intolérance. Après 3 cycles de traitement d’entretien par le lénalidomide, la dose peut être augmentée à 15 mg par voie orale en une prise par jour si elle est tolérée.
· Paliers de réduction de la posologie
|
|
Dose initiale (10 mg) |
En cas d’augmentation de la dose (15 mg)a |
|
Palier de dose -1 |
5 mg |
10 mg |
|
Palier de dose -2 |
5 mg (jours 1 à 21 de chaque cycle de 28 jours) |
5 mg |
|
Palier de dose -3 |
Sans objet |
5 mg (jours 1 à 21 de chaque cycle de 28 jours) |
|
|
Ne pas diminuer la dose en dessous de 5 mg (jours 1 à 21 de chaque cycle de 28 jours) |
|
a Après 3 cycles de lénalidomide en traitement d’entretien, la dose peut être augmentée à 15 mg par voie orale en une prise par jour si le traitement est bien toléré.
· Thrombopénie
|
Numération plaquettaire |
Action recommandée |
|
Chute < 30 x 109/L |
Interrompre le traitement par le lénalidomide |
|
Retour ≥ 30 x 109/L |
Reprendre le lénalidomide au palier de dose – 1 en une prise par jour |
|
Pour toute nouvelle rechute < 30 x 109/L |
Interrompre le traitement par le lénalidomide |
|
Retour ≥ 30 x 109/L |
Reprendre le lénalidomide à la dose immédiatement inférieure en une prise par jour |
· Numération des polynucléaires neutrophiles (PNN) - neutropénie
|
Numération des PNN |
Action recommandée a |
|
Première chute < 0,5 x 109/L |
Interrompre le traitement par le lénalidomide |
|
Retour ≥ 0,5 x 109/L |
Reprendre le lénalidomide au palier de dose – 1 en une prise par jour |
|
Pour toute nouvelle rechute < 0,5 x 109/L |
Interrompre le traitement par le lénalidomide |
|
Retour ≥ 0,5 x 109/L |
Reprendre le lénalidomide à la dose immédiatement inférieure en une prise par jour |
a À l’appréciation du médecin, si la neutropénie est la seule toxicité quel que soit le palier de dose, ajouter un facteur de croissance granulocytaire (G CSF) et maintenir la dose de lénalidomide.
· Myélome multiple chez les patients ayant reçu au moins un traitement antérieur
Le traitement par le lénalidomide ne doit pas être initié si la numération des PNN est < 1,0 x 109/L et/ou si la numération plaquettaire est < 75 x 109/L, ou, selon le niveau d’infiltration des plasmocytes dans la moelle osseuse, si la numération plaquettaire est < 30 x 109/L.
Posologie recommandée
La dose initiale recommandée est de 25 mg de lénalidomide par voie orale en une prise par jour pendant les jours 1 à 21 de chaque cycle de 28 jours. La dose recommandée de dexaméthasone est de 40 mg en une prise par jour par voie orale les jours 1 à 4, 9 à 12 et 17 à 20 de chaque cycle de 28 jours pour les 4 premiers cycles de traitement, puis de 40 mg en une prise par jour les jours 1 à 4, tous les 28 jours pour les cycles suivants.
Les médecins prescripteurs doivent déterminer avec précaution la dose de dexaméthasone à utiliser, en tenant compte de la pathologie et du statut de la maladie du patient.
· Paliers de réduction de la posologie
|
Dose initiale |
25 mg |
|
Palier de dose -1 |
15 mg |
|
Palier de dose -2 |
10 mg |
|
Palier de dose -3 |
5 mg |
· Thrombopénie
|
Numération plaquettaire |
Action recommandée |
|
Première chute < 30 x 109/L |
Interrompre le traitement par le lénalidomide |
|
Retour ≥ 30 x 109/L |
Reprendre le lénalidomide au palier de dose -1 |
|
Pour toute nouvelle rechute < 30 x 109/L |
Interrompre le traitement par le lénalidomide |
|
Retour ≥ 30 x 109/L |
Reprendre le lénalidomide au palier de dose immédiatement inférieur (palier de dose -2 ou -3) en une prise par jour. Ne pas descendre en dessous de 5 mg en une prise par jour. |
· Numération des polynucléaires neutrophiles (PNN) - neutropénie
|
Numération des PNN |
Action recommandéea |
|
Première chute < 0,5 x 109/L |
Interrompre le traitement par le lénalidomide |
|
Retour ≥ 0,5 x 109/L quand la neutropénie est la seule toxicité observée |
Reprendre le lénalidomide à la dose initiale en une prise par jour |
|
Retour ≥ 0,5 x 109/L, quand des toxicités hématologiques dépendantes de la dose autres qu’une neutropénie sont observées |
Reprendre le lénalidomide au palier de dose -1 en une prise par jour |
|
Pour toute nouvelle rechute < 0,5 x 109/L |
Interrompre le traitement par le lénalidomide |
|
Retour ≥ 0,5 x 109/L |
Reprendre le lénalidomide au palier de dose immédiatement inférieur (palier de dose -1, -2 ou -3) en une prise par jour. Ne pas descendre en dessous de 5 mg en une prise par jour. |
a À l’appréciation du médecin, si la neutropénie est la seule toxicité quel que soit le palier de dose, ajouter un facteur de croissance granulocytaire (G CSF) et maintenir la dose de lénalidomide.
· Syndromes myélodysplasiques (SMD)
Le traitement par le lénalidomide ne doit pas être initié si la numération des polynucléaires neutrophiles est < 0,5 x 109/L et/ou si la numération plaquettaire est < 25 x 109/L.
Posologie recommandée
La dose initiale recommandée est de 10 mg de lénalidomide par voie orale en une prise par jour les jours 1 à 21 de chaque cycle de 28 jours.
· Paliers de réduction de la posologie
|
Dose initiale |
10 mg en une prise par jour les jours 1 à 21 de chaque cycle de 28 jours |
|
Palier de dose -1 |
5,0 mg en une prise par jour les jours 1 à 28 de chaque cycle de 28 jours |
|
Palier de dose -2 |
2,5 mg en une prise par jour les jours 1 à 28 de chaque cycle de 28 jours |
|
Palier de dose -3 |
2,5 mg un jour sur deux les jours 1 à 28 de chaque cycle de 28 jours |
· Thrombopénie
|
Numération plaquettaire |
Action recommandée |
|
Chute < 25 x 109/L |
Interrompre le traitement par le lénalidomide |
|
Retour ≥ 25 x 109/L et < 50 x 109/L au moins à 2 reprises pendant ≥ 7 jours ou quand la récupération du taux de plaquettes est ≥ 50 x 109/L à tout moment |
Reprendre le lénalidomide au palier de dose immédiatement inférieur (palier de dose -1, -2 ou -3) |
· Numération des polynucléaires neutrophiles (PNN) - neutropénie
|
Numération des PNN |
Action recommandée |
|
Chute < 0,5 x 109/L |
Interrompre le traitement par le lénalidomide |
|
Retour ≥ 0,5 x 109/L |
Reprendre le lénalidomide au palier de dose immédiatement inférieur (palier de dose -1, -2 ou -3) |
Arrêt du traitement par le lénalidomide
Le traitement par le lénalidomide doit être arrêté chez les patients ne présentant pas au moins une réponse érythroïde mineure dans les 4 mois suivant l’instauration du traitement, démontrée par une réduction d’au moins 50 % des besoins transfusionnels ou, en l’absence de transfusions, par une augmentation de 1 g/dL du taux d’hémoglobine.
· Lymphome à cellules du manteau (LCM)
Posologie recommandée
La dose initiale recommandée est de 25 mg de lénalidomide par voie orale en une prise par jour pendant les jours 1 à 21 de chaque cycle de 28 jours.
· Paliers de réduction de la posologie
|
Dose initiale |
25 mg en une prise par jour les jours 1 à 21 de chaque cycle de 28 jours |
|
Palier de dose -1 |
20 mg en une prise par jour les jours 1 à 21 de chaque cycle de 28 jours |
|
Palier de dose -2 |
15 mg en une prise par jour les jours 1 à 21 de chaque cycle de 28 jours |
|
Palier de dose -3 |
10 mg en une prise par jour les jours 1 à 21 de chaque cycle de 28 jours |
|
Palier de dose -4 |
5 mg en une prise par jour les jours 1 à 21 de chaque cycle de 28 jours |
|
Palier de dose -5 |
2,5 mg en une prise par jour les jours 1 à 21 de chaque cycle de 28 jours1 5 mg un jour sur deux les jours 1 à 21 de chaque cycle de 28 jours |
1 Dans les pays où les gélules de 2,5 mg sont disponibles.
· Thrombopénie
|
Numération plaquettaire |
Action recommandée |
|
Chute < 50 x 109/L |
Interrompre le traitement par le lénalidomide et réaliser une numération formule sanguine (NFS) au moins tous les 7 jours |
|
Retour ≥ 60 x 109/L |
Reprendre le lénalidomide au palier de dose immédiatement inférieur (palier de dose -1) |
|
Pour toute nouvelle rechute < 50 x 109/L |
Interrompre le traitement par le lénalidomide et réaliser NFS au moins tous les 7 jours |
|
Retour ≥ 60 x 109/L |
Reprendre le lénalidomide à la dose immédiatement inférieure (palier de dose -2, -3, -4 ou -5). Ne pas descendre en dessous du palier de dose -5 |
· Numération des polynucléaires neutrophiles (PNN) - neutropénie
|
Numération des PNN |
Action recommandée |
|
Chute < 1 x 109/L pendant au moins 7 jours ou Chute < 1 x 109/L accompagnée de fièvre (température ≥ 38,5 °C) ou Chute < 0,5 x 109/L |
Interrompre le traitement par le lénalidomide et réaliser une NFS au moins tous les 7 jours |
|
Retour ≥ 1 x 109/L |
Reprendre le lénalidomide au palier de dose immédiatement inférieur (palier de dose -1) |
|
Pour toute nouvelle rechute < 1 x 109/L pendant au moins 7 jours ou rechute < 1 x 109/L accompagnée de fièvre (température ≥ 38,5°C) ou rechute < 0,5 x 109/L |
Interrompre le traitement par le lénalidomide |
|
Retour ≥ 1 x 109/L |
Reprendre le lénalidomide à la dose immédiatement inférieur (palier de dose -2, -3, -4 ou -5). Ne pas descendre en dessous du palier de dose -5 |
· Lymphome folliculaire (LF)
Le traitement par le lénalidomide ne doit pas être initié si la numération des PNN est < 1 x 109/L et/ou si la numération plaquettaire est < 50 x 109/L, à moins que ces diminutions soient secondaires à un envahissement médullaire induites par le lymphome.
Posologie recommandée
La dose initiale recommandée est de 20 mg de lénalidomide par voie orale en une prise par jour les jours 1 à 21 de chaque cycle de 28 jours, pendant 12 cycles de traitement au maximum. La dose initiale recommandée de rituximab est de 375 mg/m2 par voie intraveineuse (IV) chaque semaine au cours du cycle 1 (jours 1, 8, 15 et 22) et le jour 1 de chaque cycle de 28 jours pendant les cycles 2 à 5.
· Paliers de réduction de la posologie
|
Dose initiale |
20 mg en une prise par jour les jours 1 à 21 de chaque cycle de 28 jours |
|
Palier de dose -1 |
15 mg en une prise par jour les jours 1 à 21 de chaque cycle de 28 jours |
|
Palier de dose -2 |
10 mg en une prise par jour les jours 1 à 21 de chaque cycle de 28 jours |
|
Palier de dose -3 |
5 mg en une prise par jour les jours 1 à 21 de chaque cycle de 28 jours |
Pour les ajustements de la posologie en raison de la toxicité du rituximab, se référer au Résumé des Caractéristiques du Produit de ce médicament.
· Thrombopénie
|
Numération plaquettaire |
Action recommandée |
|
Chute < 50 x 109/L |
Interrompre le traitement par le lénalidomide et réaliser une NFS au moins tous les 7 jours |
|
Retour ≥ 50 x 109/L |
Reprendre le lénalidomide au palier de dose immédiatement inférieur (palier de dose -1) |
|
Pour toute nouvelle rechute < 50 x 109/L |
Interrompre le traitement par le lénalidomide et réaliser une NFS au moins tous les 7 jours |
|
Retour ≥ 50 x 109/L |
Reprendre le lénalidomide au palier de dose immédiatement inférieur (palier de dose -2 ou -3). Ne pas descendre en dessous du palier de dose -3 |
· Numération des polynucléaires neutrophiles (PNN) - neutropénie
|
Numération des PNN |
Action recommandéea |
|
Chute < 1 x 109/L pendant au moins 7 jours ou Chute < 1 x 109/L accompagnée de fièvre (température ≥ 38,5 °C) ou Chute < 0,5 x 109/L |
Interrompre le traitement par le lénalidomide et réaliser une NFS au moins tous les 7 jours |
|
Retour ≥ 1 x 109/L |
Reprendre le lénalidomide au palier de dose immédiatement inférieur (palier de dose -1) |
|
Pour toute nouvelle rechute < 1 x 109/L pendant au moins 7 jours ou rechute < 1 x 109/L accompagnée de fièvre (température ≥ 38,5°C) ou rechute < 0,5 x 109/L |
Interrompre le traitement par le lénalidomide et réaliser une NFS au moins tous les 7 jours |
|
Retour ≥ 1 x 109/L |
Reprendre le lénalidomide au palier de dose immédiatement inférieur (palier de dose -2 ou -3). Ne pas descendre en dessous du palier de dose -3 |
a À l’appréciation du médecin, si la neutropénie est la seule toxicité quel que soit le palier de dose, ajouter un facteur de croissance granulocytaire (G CSF).
· Lymphome à cellules du manteau (LCM) ou folliculaire (LF)
Syndrome de lyse tumorale (SLT)
Tous les patients doivent recevoir une prophylaxie du SLT (allopurinol, rasburicase ou équivalent selon le protocole utilisé dans l’établissement) et doivent être correctement hydratés (par voie orale) pendant la première semaine du premier cycle ou pendant une durée plus longue, en fonction du tableau clinique. Un bilan biochimique doit être réalisé chaque semaine pendant le premier cycle ou en fonction du tableau clinique afin de détecter la survenue d’un SLT.
Le traitement par le lénalidomide peut être poursuivi (maintien de la dose) chez les patients présentant un SLT biologique ou un SLT clinique de Grade 1, ou à l’appréciation du médecin, le traitement par le lénalidomide peut être poursuivi à une dose réduite d’un palier. Une hydratation intensive par voie intraveineuse doit être mise en place et doit s’accompagner d’une prise en charge médicale appropriée selon le protocole de soins de l’établissement, jusqu’à la correction des troubles électrolytiques. Un traitement par la rasburicase peut être nécessaire pour réduire l’hyperuricémie. La décision d’hospitalisation du patient relève de l’appréciation du médecin.
Chez les patients présentant un SLT clinique de Grades 2 à 4, le traitement par le lénalidomide doit être interrompu et un bilan biochimique doit être effectué chaque semaine ou en fonction du tableau clinique. Une hydratation intensive par voie intraveineuse doit être mise en place et doit s’accompagner d’une prise en charge médicale appropriée selon le protocole de soins de l’établissement, jusqu’à la correction des troubles électrolytiques. Le traitement par la rasburicase et la décision d’hospitalisation relèvent de l’appréciation du médecin. Après résolution du SLT (Grade 0), le traitement par le lénalidomide est repris au palier de dose immédiatement inférieur, à l’appréciation du médecin (voir rubrique 4.4).
Réaction de poussée tumorale
À l’appréciation du médecin, le traitement par le lénalidomide peut être poursuivi sans interruption ni modification chez les patients qui présentent une réaction de poussée tumorale (RPT) de Grade 1 ou 2. À l’appréciation du médecin, un traitement par anti-inflammatoires non stéroïdiens (AINS), corticoïdes d’action courte et/ou analgésiques opioïdes peut être administré. Chez les patients présentant une RPT de Grade 3 ou 4, le traitement par le lénalidomide doit être suspendu et un traitement par AINS, corticoïdes et/ou analgésiques opioïdes doit être instauré. Après résolution de la RPT à un Grade ≤ 1, reprendre le traitement par le lénalidomide à la même dose jusqu’à la fin du cycle. Un traitement symptomatique peut être administré conformément aux recommandations pour le traitement des RPT de Grades 1 et 2 (voir rubrique 4.4).
Toutes indications
En cas d’autres toxicités de Grade 3 ou 4 jugées comme étant liées au lénalidomide, le traitement doit être interrompu et ne doit être repris à la dose immédiatement inférieure que lorsque la toxicité a atteint un Grade ≤ 2 et selon l’appréciation du médecin.
Il convient d’envisager d’interrompre ou d’arrêter définitivement le traitement par le lénalidomide en cas d’éruption cutanée de Grade 2 ou 3. Le traitement par le lénalidomide doit être suspendu en cas d’angioedème, de réaction anaphylactique, d’éruption cutanée de Grade 4, d’éruption exfoliatrice ou bulleuse, ou de syndrome de Stevens-Johnson (SJS), nécrolyse épidermique toxique (NET) ou d’une réaction médicamenteuse accompagnée d’une éosinophilie et de symptômes systémiques (DRESS) et ne doit pas être repris après la résolution de ces réactions.
Populations particulières
Population pédiatrique
LENALIDOMIDE STRAGEN 10 mg, gélule ne doit pas être utilisé chez les enfants et adolescents de la naissance à moins de 18 ans en raison de problème(s) de sécurité (voir rubrique 5.1).
Sujets âgés
Les données pharmacocinétiques disponibles actuellement sont présentées à la rubrique 5.2. Lors des études cliniques, le lénalidomide a été administré chez des patients atteints de myélomes multiples et âgés jusqu’à 91 ans, chez des patients présentant un syndrome myélodysplasique et âgés jusqu’à 95 ans et chez des patients présentant un lymphome à cellules du manteau âgés jusqu’à 88 ans (voir rubrique 5.1).
Une diminution de la fonction rénale étant plus fréquente chez les patients âgés, le choix de la posologie devra être fait avec précaution chez ces patients et il est recommandé de surveiller leur fonction rénale.
· Myélome multiple non préalablement traité : patients non éligibles à une greffe
Les patients âgés de 75 ans et plus, présentant un myélome multiple non préalablement traité doivent être évalués attentivement avant d’envisager le traitement (voir rubrique 4.4).
Chez les patients âgés de plus de 75 ans traités par le lénalidomide en association avec la dexaméthasone, la dose initiale de dexaméthasone est de 20 mg par jour les jours 1, 8, 15 et 22 de chaque cycle de 28 jours.
Aucun ajustement de la posologie n’est recommandé chez les patients âgés de plus de 75 ans traités par le lénalidomide en association avec le melphalan et la prednisone.
Chez les patients âgés de 75 ans et plus, traités par le lénalidomide, présentant un myélome multiple non préalablement traité, il a été observé une incidence plus élevée d’effets indésirables graves et d’effets indésirables ayant entraîné l’arrêt du traitement.
Le traitement par le lénalidomide en association a été moins bien toléré chez les patients âgés de plus de 75 ans présentant un myélome multiple non préalablement traité, que dans la population plus jeune. Chez ces patients, le taux d’arrêts du traitement en raison d’une intolérance (événements indésirables de Grade 3 ou 4 et événements indésirables graves) a été plus élevé que chez les patients âgés de moins de 75 ans.
· Myélome multiple chez les patients ayant reçu au moins un traitement antérieur
Le pourcentage de patients âgés de 65 ans ou plus atteints d’un myélome multiple était non significativement différent dans les groupes lénalidomide/dexaméthasone et placebo/dexaméthasone. Aucune différence n’a été observée entre ces patients et les patients plus jeunes en termes de sécurité d’emploi et d’efficacité, mais on ne peut exclure une plus grande prédisposition des sujets âgés.
· Syndromes myélodysplasiques
Chez les patients atteints d’un syndrome myélodysplasique traités par le lénalidomide, aucune différence en termes de sécurité et d’efficacité n’a été observée entre les patients âgés de plus de 65 ans et les patients plus jeunes.
· Lymphome à cellules du manteau
Chez les patients atteints d’un lymphome à cellules du manteau traités par le lénalidomide, aucune différence en termes de sécurité et d’efficacité n’a été observée entre les patients âgés de plus de 65 ans et les patients de moins de 65 ans.
· Lymphome folliculaire
Chez les patients atteints d’un lymphome folliculaire traités par le lénalidomide en association avec le rituximab, le taux global des événements indésirables observé chez les patients âgés de 65 ans ou plus est comparable à celui observé chez les patients âgés de moins de 65 ans. Globalement, aucune différence en termes d’efficacité n’a été observée entre les deux tranches d’âge.
Patients présentant une insuffisance rénale
Le lénalidomide est essentiellement excrété par les reins ; la tolérance du traitement peut être diminuée chez les patients présentant un degré élevé d’insuffisance rénale (voir rubrique 4.4). Le choix de la posologie devra être fait avec précaution et il est recommandé que la fonction rénale soit surveillée.
Aucun ajustement de la posologie n’est nécessaire chez les patients atteints d’un myélome multiple ou d’un lymphome folliculaire présentant une insuffisance rénale légère.
Les ajustements de posologie suivants sont recommandés en début de traitement et pendant tout le traitement en cas d’insuffisance rénale modérée ou sévère ou en cas d’insuffisance rénale terminale. Il n’existe pas de données d’étude de phase 3 chez les patients atteints d’insuffisance rénale terminale (IRT) (ClCr < 30 mL/min, nécessitant une dialyse).
· Myélome multiple
|
Fonction rénale (ClCr) |
Ajustement de la posologie
|
|
Insuffisance rénale modérée (30 ≤ ClCr < 50 mL/min) |
10 mg en une prise par jour1 |
|
Insuffisance rénale sévère (ClCr < 30 mL/min, ne nécessitant pas de dialyse) |
7,5 mg en une prise par jour2 15 mg un jour sur deux |
|
Insuffisance rénale terminale (IRT) (ClCr < 30 mL/min, nécessitant une dialyse) |
5 mg en une prise par jour. Les jours de dialyse, la dose doit être administrée après la dialyse. |
1 La dose pourra être augmentée à 15 mg en une prise par jour au bout de 2 cycles si le patient ne répond pas au traitement et s’il le tolère.
2 Dans les pays où les gélules de 7,5 mg sont commercialisées.
· Syndromes myélodysplasiques
|
Fonction rénale (ClCr) |
Ajustement de la posologie |
|
|
Insuffisance rénale modérée (30 ≤ ClCr < 50 mL/min) |
Dose initiale |
5 mg en une prise par jour (jours 1 à 21 de chaque cycle de 28 jours) |
|
Palier de dose -1* |
2,5 mg en une prise par jour (jours 1 à 28 de chaque cycle de 28 jours) |
|
|
Palier de dose -2* |
2,5 mg tous les 2 jours (jours 1 à 28 de chaque cycle de 28 jours) |
|
|
Insuffisance rénale sévère (ClCr < 30 mL/min, ne nécessitant pas de dialyse) |
Dose initiale |
2,5 mg en une prise par jour (jours 1 à 21 de chaque cycle de 28 jours) |
|
Palier de dose -1* |
2,5 mg tous les 2 jours (jours 1 à 28 de chaque cycle de 28 jours) |
|
|
Palier de dose -2* |
2,5 mg deux fois par semaine (jours 1 à 28 de chaque cycle de 28 jours) |
|
|
Insuffisance rénale terminale (IRT) (ClCr < 30 mL/min, nécessitant une dialyse)
Les jours de dialyse, la dose doit être administrée après la dialyse. |
Dose initiale |
2,5 mg en une prise par jour (jours 1 à 21 de chaque cycle de 28 jours) |
|
Palier de dose -1* |
2,5 mg tous les 2 jours (jours 1 à 28 de chaque cycle de 28 jours) |
|
|
Palier de dose -2* |
2,5 mg deux fois par semaine (jours 1 à 28 de chaque cycle de 28 jours) |
|
* Paliers de réduction de la posologie recommandés en cours de traitement et lors de la reprise du traitement pour prendre en charge les thrombopénies ou neutropénies de Grade 3 ou 4, ou autres effets toxiques de Grade 3 ou 4 jugés en rapport avec le lénalidomide, tels que présentés ci-dessus.
· Lymphome à cellules du manteau
|
Fonction rénale (ClCr) |
Ajustement de la posologie (jours 1 à 21 de chaque cycle de 28 jours) |
|
Insuffisance rénale modérée (30 ≤ ClCr < 50 mL/min) |
10 mg en une prise par jour1 |
|
Insuffisance rénale sévère (ClCr < 30 mL/min, ne nécessitant pas de dialyse) |
7,5 mg en une prise par jour2 15 mg un jour sur deux |
|
Insuffisance rénale terminale (IRT) (ClCr < 30 mL/min, nécessitant une dialyse) |
5 mg en une prise par jour. Les jours de dialyse, la dose doit être administrée après la dialyse |
1 La dose pourra être augmentée à 15 mg en une prise par jour au bout de 2 cycles si le patient ne répond pas au traitement et s’il le tolère.
2 Dans les pays où les gélules de 7,5 mg sont commercialisées.
· Lymphome folliculaire
|
Fonction rénale (ClCr) |
Ajustement de la posologie (Jours 1 à 21 de chaque cycle de 28 jours) |
|
Insuffisance rénale modérée (30 ≤ ClCr < 60 mL/min) |
10 mg en une prise par jour1,2 |
|
Insuffisance rénale sévère (ClCr < 30 mL/min, ne nécessitant pas de dialyse) |
5 mg en une prise par jour |
|
Insuffisance rénale terminale (IRT) (ClCr < 30 mL/min, nécessitant une dialyse) |
5 mg en une prise par jour. Les jours de dialyse, la dose doit être administrée après la dialyse |
1 La dose pourra être augmentée à 15 mg en une prise par jour au bout de 2 cycles si le patient tolère le traitement.
2 Chez les patients recevant une dose initiale de 10 mg, en cas de diminution de la dose dans le cadre de la prise en charge d’une neutropénie ou d’une thrombopénie de Grade 3 ou 4, ou autres effets toxiques de Grade 3 ou 4 jugés en rapport avec le lénalidomide, ne pas administrer une dose inférieure à 5 mg un jour sur deux ou 2,5 mg en une prise par jour.
Après l’instauration du traitement par le lénalidomide, toute modification ultérieure de la dose de lénalidomide chez les patients présentant une insuffisance rénale doit reposer sur la tolérance au traitement de chaque individu, telle que décrite ci-dessus.
Patients présentant une insuffisance hépatique
L’utilisation du lénalidomide chez le sujet présentant une insuffisance hépatique n’a pas fait l’objet d’études formelles et aucune adaptation de la posologie particulière ne peut être recommandée.
Mode d’administration
Voie orale.
Les gélules de LENALIDOMIDE STRAGEN 10 mg, doivent être prises par voie orale environ à la même heure les jours prévus de chaque cycle. Les gélules ne doivent pas être ouvertes, cassées ou mâchées. Les gélules doivent être avalées entières, de préférence avec de l’eau, au cours ou en dehors des repas.
Il est recommandé d’appuyer sur une extrémité seulement de la gélule pour l’extraire de la plaquette, ce qui réduit le risque de déformation ou de rupture de la gélule.
· Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1.
· Femmes enceintes.
· Femmes en âge de procréer, à moins que toutes les conditions requises par le Programme de Prévention de la Grossesse soient remplies (voir rubriques 4.4 et 4.6).
4.4. Mises en garde spéciales et précautions d'emploi
Mise en garde relative à la grossesse
Le lénalidomide est structurellement proche du thalidomide. Le thalidomide est une substance active tératogène humaine connue, provoquant des anomalies congénitales graves, potentiellement létales chez l’enfant à naître. Le lénalidomide induit chez les singes des malformations similaires à celles décrites avec le thalidomide (voir rubriques 4.6 et 5.3). Si le lénalidomide est pris pendant la grossesse, un effet tératogène du lénalidomide est attendu chez l’être humain.
Les conditions requises par le Programme de Prévention de la Grossesse doivent être complètement remplies par toutes les patientes, à moins de pouvoir affirmer avec certitude que la patiente est dans l’impossibilité de procréer.
Critères permettant de définir qu’une femme est dans l’impossibilité de procréer
Toute patiente ou partenaire de patient est considérée comme en âge de procréer, sauf si elle présente au moins l’un des critères suivants :
· Age ≥ 50 ans et aménorrhée naturelle depuis au moins 1 an (l’aménorrhée faisant suite au traitement d’un cancer ou pendant l’allaitement ne suffit pas à exclure un risque de grossesse)
· Ménopause précoce confirmée par un gynécologue spécialisé
· Salpingo-ovariectomie bilatérale ou hystérectomie antérieures
· Génotype XY, syndrome de Turner, agénésie utérine
Information des patients
Pour les femmes en âge de procréer, le lénalidomide est contre-indiqué à moins que tous les éléments suivants soient respectés :
· Elle comprend le risque tératogène attendu pour l’enfant à naître
· Elle comprend la nécessité d’une contraception efficace, sans interruption, commencée au moins 4 semaines avant le début du traitement, poursuivie pendant toute la durée du traitement, et pendant au moins 4 semaines après la fin du traitement
· Même si une femme en âge de procréer souffre potentiellement d’une aménorrhée, elle doit suivre tous les conseils sur la contraception efficace
· Elle doit être capable de se conformer à des mesures contraceptives efficaces
· Elle est informée et elle comprend les conséquences potentielles d’une grossesse et la nécessité de consulter rapidement s’il y a un risque de grossesse
· Elle comprend la nécessité de débuter le traitement dès que le lénalidomide est délivré à la suite d’un test de grossesse négatif
· Elle comprend la nécessité et accepte de subir un test de grossesse au moins toutes les 4 semaines sauf en cas de stérilisation tubaire confirmée
· Elle reconnaît qu’elle comprend les dangers et les précautions nécessaires associés à l’utilisation du lénalidomide
Pour les hommes traités par le lénalidomide, les données pharmacocinétiques ont montré que le lénalidomide était présent dans le sperme humain à des concentrations extrêmement faibles pendant le traitement et qu’il était indétectable 3 jours après l’arrêt du médicament chez les sujets sains (voir rubrique 5.2). À titre de précaution et en tenant compte de l’allongement du temps d’élimination dans les populations particulières telles que les patients présentant une insuffisance rénale, tous les hommes traités par le lénalidomide doivent :
· Comprendre les risques tératogènes attendus en cas de rapport sexuel avec une femme enceinte ou avec une femme en âge de procréer
· Comprendre la nécessité d’utiliser un préservatif en cas de rapport sexuel avec une femme enceinte ou une femme en âge de procréer n’utilisant pas une contraception efficace (même si le patient a subi une vasectomie), pendant le traitement et pendant au moins 7 jours après l’interruption et/ou l’arrêt du traitement.
· Comprendre qu’en cas de survenue d’une grossesse chez leur partenaire pendant qu’il prend LENALIDOMIDE STRAGEN 10 mg, gélule ou peu après l’arrêt de la prise de LENALIDOMIDE STRAGEN 10 mg, gélule il doit informer immédiatement leur médecin traitant et qu’il est recommandé d’adresser la partenaire à un médecin spécialiste ou expérimenté en tératologie pour évaluation et conseil
Le médecin prescripteur doit s’assurer que pour les femmes en âge de procréer :
· La patiente remplit les conditions requises par le Programme de Prévention de la Grossesse, y compris la confirmation qu'elle dispose d’un niveau approprié de compréhension
· La patiente confirme avoir compris les conditions susmentionnées
Contraception
Les femmes en âge de procréer doivent utiliser au moins une méthode de contraception efficace pendant au moins 4 semaines avant le début du traitement, pendant le traitement, et pendant au moins 4 semaines après l’arrêt du traitement par le lénalidomide, même en cas d’interruption du traitement à moins que la patiente déclare une abstinence totale et continue confirmée de façon mensuelle. Si la patiente n’utilise aucun moyen de contraception efficace, elle devra être orientée vers un médecin compétent pour être conseillée sur la contraception de façon à ce qu’une contraception soit instaurée.
Les exemples suivants sont considérés comme des exemples de méthodes de contraception adaptées :
· Implant contraceptif
· Dispositif Intra-Utérin (DIU) au lévonorgestrel
· Acétate de médroxyprogestérone-retard
· Stérilisation tubaire
· Rapports sexuels exclusivement avec un partenaire vasectomisé ; la vasectomie doit avoir été confirmée par deux spermogrammes négatifs
· Pilule progestative inhibant l’ovulation (c’est-à-dire désogestrel)
En raison du risque accru d’accidents thrombo-emboliques veineux chez les patients atteints de myélome multiple et traités par le lénalidomide en association et, dans une moindre mesure, chez les patients présentant un myélome multiple, un syndrome myélodysplasique ou un lymphome à cellules du manteau traités par le lénalidomide en monothérapie, l’utilisation de pilules contraceptives orales combinées n’est pas recommandée (voir également rubrique 4.5). Si la patiente est sous contraceptif oral combiné, elle devra utiliser une des autres méthodes contraceptives efficaces citées ci-dessus. Le risque thrombo-embolique persiste pendant 4 à 6 semaines après l’arrêt du contraceptif oral combiné. L’efficacité des contraceptifs stéroïdiens peut être diminuée en cas de traitement concomitant par la dexaméthasone (voir rubrique 4.5).
Les implants contraceptifs et les dispositifs intra-utérins au lévonorgestrel sont associés à un risque accru d’infection lors de leur insertion et à des saignements vaginaux irréguliers. Le recours aux antibiotiques à titre prophylactique devra être envisagé lors de leur mise en place, en particulier en cas de neutropénie associée.
L’utilisation de dispositifs intra-utérins au cuivre n’est de manière générale pas recommandée en raison des risques d’infection lors de leur insertion et des règles abondantes qu’elle peut entraîner, susceptibles de mettre en danger les patientes présentant une neutropénie ou une thrombopénie.
Test de grossesse
Selon la pratique locale, les tests de grossesse avec une sensibilité minimale de 25 mIU/mL doivent être effectués sous contrôle médical pour les femmes en âge de procréer comme indiqué ci-dessous. Cette obligation inclue également les femmes en âge de procréer qui pratiquent l'abstinence absolue et continue. Idéalement, le test de grossesse, la prescription et la délivrance auront lieu le même jour. La délivrance du lénalidomide aux femmes en âge de procréer devrait avoir lieu dans les 7 jours suivant la prescription.
Avant de commencer le traitement
Un test de grossesse doit être fait sous contrôle médical lors de la consultation, quand le lénalidomide est prescrit, ou dans les 3 jours précédant la consultation si la patiente utilise une contraception efficace depuis au moins 4 semaines. Le test doit confirmer que la patiente n’est pas enceinte au moment où elle débute le traitement par le lénalidomide.
Suivi et arrêt du traitement
Un nouveau test de grossesse sous contrôle médical doit être effectué au moins toutes les 4 semaines et pendant au moins 4 semaines après l’arrêt du traitement, sauf en cas de stérilisation tubaire confirmée. Ces tests de grossesse doivent être faits le jour de la consultation dédiée à la prescription ou dans les 3 jours précédents la consultation.
Précautions supplémentaires
Les patients doivent être informés de ne jamais donner leur médicament à quelqu’un d’autre et de rapporter les gélules non utilisées à leur pharmacien en fin de traitement pour une élimination en toute sécurité.
Les patients ne doivent pas faire de don de sang ou de sperme pendant la prise de lénalidomide (y compris pendant les interruptions de traitement) et pendant au moins 7 jours après la fin du traitement.
Les professionnels de santé et les aidants doivent porter des gants jetables pour manipuler la plaquette ou la gélule. Les femmes enceintes ou qui pensent l’être ne doivent pas manipuler la plaquette ou la gélule (voir rubrique 6.6).
Guide d’aide à la prescription, restrictions de prescription et de délivrance
Afin d’aider les patients à éviter toute exposition fœtale au lénalidomide, le titulaire de l’autorisation de mise sur le marché (AMM) fournira aux professionnels de santé des documents explicatifs visant à renforcer les mises en garde relatives à la tératogénicité attendue du lénalidomide, à donner des conseils pour la mise en place d’une contraception préalable au traitement et à fournir des explications sur les tests de grossesse nécessaires. Le prescripteur doit informer le patient du risque tératogène attendu et des mesures contraceptives strictes définies dans le programme de prévention de la grossesse et lui remettre la brochure appropriée d’information destinée aux patients, la carte patient et/ou un document équivalent, comme défini avec chaque autorité compétente nationale. En collaboration avec chaque autorité compétente nationale, un programme d’accès contrôlé a été mis en place qui inclut l’utilisation d’une carte patient et/ou d’un document équivalent pour le contrôle des prescriptions et/ou des délivrances et le recueil d’information relatives à l’indication afin de surveiller l’utilisation hors AMM sur le territoire national. Dans l’idéal, le test de grossesse, la prescription et la délivrance du médicament doivent avoir lieu le même jour. La délivrance du lénalidomide aux femmes en âge de procréer doit avoir lieu dans les 7 jours suivant la prescription et un test de grossesse négatif effectué sous contrôle médical. La prescription doit être limitée à 4 semaines de traitement au maximum conformément aux schémas posologiques dans les indications autorisées (voir rubrique 4.2) chez les femmes en âge de procréer et à 12 semaines au maximum chez tous les autres patients.
Autres mises en garde spéciales et précautions d’emploi
Infarctus du myocarde
Des cas d’infarctus du myocarde ont été rapportés chez les patients recevant du lénalidomide, particulièrement chez ceux qui présentent des facteurs de risque connus et au cours des 12 premiers mois de traitement lorsque le lénalidomide est utilisé en association avec la dexaméthasone. Les patients présentant des facteurs de risque connus – y compris un antécédent de thrombose – doivent être surveillés étroitement, et des mesures doivent être prises pour essayer de réduire au minimum tous les facteurs de risque modifiables (par exemple le tabagisme, l’hypertension et l’hyperlipidémie).
Événements thrombo-emboliques artériels et veineux
Chez les patients atteints de myélome multiple, l’utilisation concomitante du lénalidomide et de la dexaméthasone est associée à un risque accru de thrombo-embolie veineuse (principalement thrombose veineuse profonde et embolie pulmonaire). Le risque de thrombo-embolie veineuse a été observé dans une moindre mesure avec le lénalidomide en association avec le melphalan et la prednisone.
Chez les patients présentant un myélome multiple, un syndrome myélodysplasique ou un lymphome à cellules du manteau, le traitement par le lénalidomide en monothérapie est associé à un risque plus faible de thrombo-embolie veineuse (principalement thrombose veineuse profonde et embolie pulmonaire) que chez les patients atteints d’un myélome multiple traités par lénalidomide en association (voir rubriques 4.5 et 4.8).
Chez les patients atteints d'un myélome multiple, l’utilisation concomitante du lénalidomide et de la dexaméthasone est associée à un risque accru de thrombo-embolie artérielle (principalement infarctus du myocarde et accident vasculaire cérébral) qui a été observé dans une moindre mesure avec le lénalidomide administré en association avec le melphalan et la prednisone. Le risque de thrombo-embolie artérielle est plus faible chez les patients atteints de myélome multiple traités par le lénalidomide en monothérapie que chez les patients traités par le lénalidomide en association.
Par conséquent, les patients présentant des facteurs de risque connus – y compris un antécédent de thrombose – doivent être surveillés étroitement. Des mesures doivent être prises pour essayer de réduire au minimum tous les facteurs de risque modifiables (par exemple le tabagisme, l’hypertension et l’hyperlipidémie). L’administration concomitante d’érythropoïétine ou des antécédents d’événements thrombo-emboliques peuvent également augmenter les risques de thrombose veineuse chez ces patients. Par conséquent, l’érythropoïétine et les autres médicaments pouvant accroître les risques de thrombose, comme les traitements hormonaux substitutifs, doivent être utilisés avec précaution chez les patients atteints de myélome multiple traités par le lénalidomide et la dexaméthasone. Un taux d’hémoglobine supérieur à 12 g/dL doit conduire à l’arrêt de l’érythropoïétine.
Il est conseillé aux patients et à leurs médecins d’être attentifs aux signes et symptômes de thrombo-embolie. Il doit être recommandé aux patients de consulter un médecin en cas de survenue de symptômes tels qu’essoufflement, douleur thoracique, gonflement des bras ou jambes. La prescription d’antithrombotiques en prophylaxie est recommandée, en particulier chez les patients présentant des facteurs de risque de thrombose supplémentaires. La décision de mettre en place des mesures prophylactiques antithrombotiques devra être prise au cas par cas en fonction des facteurs de risque sous-jacents propres à chaque patient.
Si le patient ressent des événements thrombo-emboliques le traitement doit être interrompu et une thérapie anticoagulante standard doit être mise en œuvre. Une fois le patient stabilisé sous traitement anticoagulant et toute complication éventuelle de l’événement thrombo-embolique écartée, le traitement par le lénalidomide peut être repris à la dose d’origine, en fonction de l’évaluation du rapport bénéfice-risque. Le patient doit poursuivre son traitement anticoagulant pendant toute la durée du traitement par le lénalidomide.
Hypertension pulmonaire
Des cas d’hypertension pulmonaire, parfois d’issue fatale, ont été rapportés chez des patients traités par le lénalidomide. Les signes et symptômes de maladie cardiopulmonaire sous-jacente doivent être évalués avant l’instauration du traitement et pendant le traitement par le lénalidomide.
Neutropénie et thrombopénie
Les principales toxicités dose-limitantes du lénalidomide sont la neutropénie et la thrombopénie. Un hémogramme complet, avec formule leucocytaire, numération plaquettaire, hémoglobine et hématocrite, doit être réalisé avant le traitement, une fois par semaine pendant les 8 premières semaines de traitement par le lénalidomide, puis une fois par mois pour surveiller l’apparition de cytopénies. Chez les patients présentant un lymphome à cellules du manteau, les contrôles doivent être réalisés toutes les deux semaines pendant les cycles 3 et 4, puis au début de chaque cycle. Chez les patients atteints d’un lymphome folliculaire, les contrôles doivent être réalisés une fois par semaine pendant les trois premières semaines du cycle 1 (28 jours), toutes les deux semaines au cours des cycles 2 à 4, puis au début de chaque cycle suivant. Une interruption du traitement et/ou une réduction de la dose peut s’avérer nécessaire (voir rubrique 4.2).
En cas de neutropénie, le médecin devra envisager l’utilisation de facteurs de croissance dans la prise en charge du patient. Les patients doivent être informés de la nécessité de signaler rapidement tout épisode fébrile.
Il est conseillé aux patients et à leurs médecins d’être attentifs aux signes et symptômes évocateurs d’une hémorragie, y compris les pétéchies et épistaxis, notamment chez les patients recevant un traitement concomitant susceptible d’induire des saignements (voir rubrique 4.8 Troubles hémorragiques).
L’administration concomitante de lénalidomide avec d’autres myélosuppresseurs doit être entreprise avec précaution.
· Myélome multiple non préalablement traité : patients recevant le lénalidomide en traitement d’entretien après une AGCS
Les effets indésirables observés dans l'étude CALGB 100104 incluaient les événements signalés après un traitement par melphalan à forte dose et une AGCS (MFD/AGCS), ainsi que les événements survenus au cours de la période de traitement d'entretien. Une deuxième analyse a permis d’identifier les événements survenus après le début du traitement d’entretien. Dans l'étude IFM 2005-02, les effets indésirables ne concernaient que la période de traitement d'entretien.
Globalement, les neutropénies de Grade 4 ont été observées à une fréquence plus élevée dans les bras lénalidomide en traitement d’entretien que dans les bras placebo en traitement d’entretien dans les 2 études évaluant le lénalidomide en traitement d’entretien chez des patients atteints d’un myélome multiple non préalablement traité ayant reçu une AGCS (32,1 % contre 26,7 % [16,1 % contre 1,8 % après le début du traitement d’entretien] dans l'étude CALGB 100104 et 16,4 % contre 0,7 % dans l'étude IFM 2005-02, respectivement). Des effets indésirables de type neutropénie apparus sous traitement entraînant l’arrêt du lénalidomide ont été rapportés chez 2,2 % des patients dans l'étude CALGB 100104 et 2,4 % des patients dans l'étude IFM 2005-02, respectivement. Dans les deux études, des neutropénies fébriles de Grade 4 ont été rapportées avec des fréquences similaires dans les bras lénalidomide en traitement d’entretien et dans les bras placebo en traitement d’entretien (0,4 % contre 0,5 % [0,4 % contre 0,5 % après le début du traitement d’entretien] dans l'étude CALGB 100104 et 0,3 % contre 0 % dans l'étude IFM 2005-02, respectivement). Les patients doivent être informés de la nécessité de signaler rapidement tout épisode fébrile, une interruption du traitement et/ou des réductions de la dose peuvent être nécessaires (voir rubrique 4.2).
Des thrombopénies de Grades 3 et 4 ont été observées à une fréquence plus élevée dans les bras lénalidomide en traitement d’entretien que dans les bras placebo dans les études évaluant le lénalidomide en traitement d’entretien chez des patients atteints d’un myélome multiple non préalablement traité ayant reçu une AGCS (37,5 % contre 30,3 % [17,9 % contre 4,1 % après le début du traitement d’entretien] dans l'étude CALGB 100104 et 13,0 % contre 2,9 % dans l'étude IFM 2005-02, respectivement). Il est conseillé aux patients et à leurs médecins d’être attentifs aux signes et symptômes évocateurs d’une hémorragie, y compris les pétéchies et l'épistaxis, notamment chez les patients recevant un traitement concomitant susceptible d’induire des saignements (voir rubrique 4.8, Troubles hémorragiques).
La fréquence des neutropénies de Grade 4 a été plus faible dans le bras lénalidomide en association avec le bortézomib et la dexaméthasone (RVd) que dans le bras comparateur Rd (2,7 % versus 5,9 %) de l’étude SWOG SO777. La fréquence des neutropénies fébriles de Grade 4 a été comparable dans le bras RVd et dans le bras Rd (0,0 % versus 0,4 %). Les patients doivent être informés de la nécessité de signaler rapidement tout épisode fébrile ; une interruption du traitement et/ou une réduction de la dose peuvent être nécessaires (voir rubrique 4.2).
La fréquence des thrombopénies de Grade 3 ou 4 a été plus élevée dans le bras RVd que dans le bras comparateur Rd (17,2 % versus 9,4 %).
· Myélome multiple non préalablement traité : patients, non éligibles à une greffe, traités par le lénalidomide en association avec la dexaméthasone à faible dose
Des neutropénies de Grade 4 ont été observées à une fréquence plus faible dans les bras lénalidomide en association avec la dexaméthasone que dans le bras comparateur (8,5 % dans les Rd [traitement continu] et Rd18 [traitement pendant 18 cycles de 4 semaines] contre 15 % dans le bras melphalan/prednisone/thalidomide, voir rubrique 4.8). Des épisodes de neutropénie fébrile de Grade 4 étaient cohérents à ceux observés dans le bras comparateur (0,6 % dans les Rd et Rd18 chez les patients traités par lénalidomide/dexaméthasone contre 0,7 % dans le bras melphalan/prednisone/thalidomide, voir rubrique 4.8).
Des thrombopénies de Grade 3 ou 4 ont été observées à une plus faible mesure dans les bras Rd et Rd18 que dans le bras comparateur (8,1 % contre 11,1 %, respectivement).
· Myélome multiple non préalablement traité : patients non éligibles à une greffe, traités par le lénalidomide en association avec le melphalan et la prednisone
L’association du lénalidomide avec le melphalan et la prednisone dans les études cliniques menées chez les patients atteints d’un myélome multiple non préalablement traité, est associée à une fréquence accrue de neutropénies de Grade 4 (34,1 % chez les patients traités par le bras melphalan, prednisone et lénalidomide suivis du lénalidomide [MPR+R] et melphalan, prednisone et lénalidomide suivis du placebo [MPR+p] contre 7,8 % chez les patients traités par MPp+p; voir rubrique 4.8). Des épisodes de neutropénie fébrile de Grade 4 ont été observés peu fréquemment (1,7 % chez les patients traités par MPR+R/MPR+p contre 0.0 % chez les patients traités par MPp+p ; voir rubrique 4.8).
L’association du lénalidomide avec le melphalan et la prednisone chez les patients atteints d’un myélome multiple non préalablement traité est associée à une fréquence accrue de thrombopénies de Grade 3 et 4 (40,4 % chez les patients traités par MPR+R/MPR+p contre 13,7 % chez les patients traités par MPp+p ; voir rubrique 4.8).
· Myélome multiple : patients ayant reçu au moins un traitement antérieur
L’association du lénalidomide et de la dexaméthasone chez les patients atteints de myélomes multiples ayant reçu au moins un traitement antérieur est associée à une incidence accrue des neutropénies de Grade 4 (5,1 % des patients traités par lénalidomide/dexaméthasone contre 0,6 % des patients traités par placebo/dexaméthasone; voir rubrique 4.8). Des épisodes neutropéniques fébriles de Grade 4 ont plus rarement été observés (0,6 % des patients traités par lénalidomide/dexaméthasone contre 0,0 % des patients traités par placebo/dexaméthasone; voir rubrique 4.8).
L’association du lénalidomide et de la dexaméthasone chez les patients atteints de myélome multiple est associée à une incidence accrue des thrombopénies de Grade 3 et de Grade 4 (respectivement 9,9 % et 1,4 % chez les patients traités par lénalidomide/dexaméthasone contre 2,3 % et 0,0 % chez les patients traités par placebo/dexaméthasone; voir rubrique 4.8).
· Syndromes myélodysplasiques
Chez les patients présentant un syndrome myélodysplasique, le traitement par le lénalidomide est associé à une fréquence accrue de neutropénie et thrombopénie de Grade 3 et 4 par rapport aux patients recevant du placebo (voir rubrique 4.8).
· Lymphome à cellules du manteau
Chez les patients présentant un lymphome à cellules du manteau, le traitement par le lénalidomide est associé à une fréquence accrue de neutropénie de Grades 3 et 4 par rapport aux patients du bras contrôle (voir rubrique 4.8).
· Lymphome folliculaire
L’association du lénalidomide et du rituximab chez les patients atteints de lymphome folliculaire est associée à une incidence plus élevée de neutropénies de Grade 3 ou 4 par rapport aux patients du bras placebo/rituximab. Des neutropénies fébriles et des thrombopénies de Grade 3 ou 4 ont été observées plus fréquemment dans le bras lénalidomide/rituximab (voir rubrique 4.8).
Affections thyroïdiennes
Des cas d’hypothyroïdie et des cas d’hyperthyroïdie ont été rapportés. Un contrôle optimal des conditions de comorbidités influençant la fonction thyroïdienne est recommandé avant l’instauration du traitement. Un contrôle de la fonction thyroïdienne est recommandé avant le début du traitement et régulièrement ensuite.
Neuropathie périphérique
Le lénalidomide est structurellement proche du thalidomide, qui est connu pour entraîner des neuropathies périphériques sévères. Il n’a pas été observé d’augmentation de la fréquence de neuropathie périphérique lors de l’administration de lénalidomide en association avec la dexaméthasone ou avec le melphalan et la prednisone, en monothérapie ou lors de l’administration au long cours de lénalidomide dans le traitement du myélome multiple non préalablement traité.
Une fréquence plus élevée de neuropathies périphériques est observée avec l’association de lénalidomide et de bortézomib intraveineux et dexaméthasone chez les patients présentant un myélome multiple. La fréquence était plus faible lorsque le bortézomib était administré par voie sous-cutanée. Pour des informations supplémentaires, voir la rubrique 4.8 et le Résumé des Caractéristiques du Produit du bortézomib.
Réaction de poussée tumorale et syndrome de lyse tumorale
Le lénalidomide ayant une activité antinéoplasique, des complications de type syndrome de lyse tumorale (SLT) peuvent survenir. Des cas de SLT et de réaction de poussée tumorale (RPT), parfois d’issue fatale, ont été rapportés (voir rubrique 4.8). Les patients présentant des risques de SLT et de RPT sont ceux qui ont une charge tumorale élevée avant le traitement.
Le traitement par le lénalidomide doit être instauré avec prudence chez ces patients. Ces patients doivent faire l’objet d’une surveillance étroite, en particulier pendant le premier cycle ou après une augmentation de la dose, et les précautions appropriées doivent être prises.
· Lymphome à cellules du manteau
Une surveillance étroite et une évaluation du patient afin de détecter une RPT sont recommandées. Les patients ayant un score MIPI (Mantle Cell International Prognostic Index) élevé lors du diagnostic ou une charge tumorale élevée (au moins une lésion mesurant ≥ 7 cm dans le plus grand diamètre) avant l’initiation du traitement peuvent présenter un risque de RPT. La réaction de poussée tumorale peut simuler une progression de la maladie (MP – maladie en progression). Dans les études MCL-002 et MCL-001, les patients qui présentaient une RPT de Grade 1 ou 2 recevaient un traitement symptomatique de la RPT par des corticoïdes, des anti-inflammatoires non stéroïdiens (AINS) et/ou des analgésiques opioïdes. La décision d’instaurer des mesures thérapeutiques pour la RPT doit être prise après une évaluation clinique attentive de chaque patient (voir rubriques 4.2 et 4.8).
· Lymphome folliculaire
Une surveillance étroite et une évaluation du patient afin de détecter une RPT sont recommandées. La réaction de poussée tumorale peut simuler une progression de la maladie. Les patients qui présentaient une RPT de Grade 1 ou 2 recevaient un traitement symptomatique de la RPT par des corticoïdes, des AINS et/ou des analgésiques opioïdes. La décision d’instaurer des mesures thérapeutiques pour la RPT doit être prise après une évaluation clinique attentive de chaque patient (voir rubriques 4.2 et 4.8).
Une surveillance étroite et une évaluation du patient afin de détecter un SLT sont recommandées. Les patients doivent être correctement hydratés et doivent recevoir une prophylaxie du SLT en plus du bilan biochimique hebdomadaire pendant le premier cycle ou plus longtemps, en fonction du tableau clinique (voir rubriques 4.2 et 4.8).
Charge tumorale
· Lymphome à cellules du manteau
Le lénalidomide n’est pas recommandé dans le traitement des patients ayant une charge tumorale élevée si d’autres options thérapeutiques sont disponibles.
Décès prématurés
Dans l’étude MCL-002, il a été observé globalement une augmentation apparente des décès prématurés (au cours des 20 premières semaines). Les patients ayant une charge tumorale élevée avant l’initiation du traitement présentent un risque accru de décès prématuré : des décès prématurés ont été rapportés chez 16/81 patients (20 %) du bras lénalidomide et 2/28 patients (7 %) du bras contrôle. Les chiffres correspondants sur 52 semaines étaient de 32/81 patients (40 %) et 6/28 patients (21 %) (voir rubrique 5.1).
Événements indésirables
Dans l’étude MCL-002, pendant le cycle 1 de traitement, le traitement a été arrêté chez 11/81 patients (14 %) ayant une charge tumorale élevée du bras lénalidomide versus 1/28 patients (4 %) du bras contrôle. La survenue d’événements indésirables était la principale raison d’arrêt du traitement pendant le cycle 1 chez les patients ayant une charge tumorale élevée (7/11, 64 %).
Les patients ayant une charge tumorale élevée doivent donc être surveillés étroitement pour détecter l’apparition d’effets indésirables (voir rubrique 4.8), y compris des signes de réaction de poussée tumorale (RPT). Se reporter à la rubrique 4.2 pour les ajustements de la posologie en cas de RPT. Une charge tumorale élevée était définie comme la présence d’au moins une lésion mesurant ≥ 5 cm de diamètre ou d’au moins 3 lésions mesurant ≥ 3 cm.
Réactions allergiques et réactions cutanées sévères
Des cas de réactions allergiques, y compris d’angio-œdème, de réaction anaphylactique, et de réactions cutanées sévères, comme le syndrome de Stevens-Johnson (SJS), la nécrolyse épidermique toxique (NET) et la réaction médicamenteuse accompagnée d’une éosinophilie et de symptômes systémiques (syndrome DRESS), ont été rapportées chez des patients traités par le lénalidomide (voir rubrique 4.8).
Les patients doivent être informés des signes et symptômes de ces réactions par leur médecin et avertis de la nécessité de consulter immédiatement un médecin s’ils présentent ces symptômes. Le traitement par le lénalidomide doit être arrêté en cas d’angio-œdème, de réaction anaphylactique, d’éruption exfoliatrice ou bulleuse ou en cas de suspicion de SJS, de NET ou de syndrome DRESS, et ne doit pas être repris après la résolution de ces réactions. En cas d’autres formes de réaction cutanée, une interruption ou l’arrêt du traitement par le lénalidomide en fonction de la sévérité de la réaction doivent être envisagés. Les patients ayant présenté des réactions allergiques pendant un traitement antérieur par le thalidomide doivent être surveillés étroitement car la possibilité d’une réaction croisée entre le lénalidomide et le thalidomide a été rapportée dans la littérature. Les patients ayant des antécédents d’éruption sévère associée au traitement par le thalidomide ne doivent pas recevoir le lénalidomide.
Intolérance au lactose
Les gélules de LENALIDOMIDE STRAGEN 10 mg contiennent du lactose. Les patients présentant des problèmes héréditaires rares d’intolérance au galactose, un déficit total en lactase ou un syndrome de malabsorption du glucose et du galactose ne doivent pas prendre ce médicament.
Cancers secondaires au traitement
Une augmentation de l’incidence de cancers secondaires (CS) a été observée dans les études cliniques menées chez des patients sous lénalidomide/dexaméthasone ayant déjà reçu un traitement pour leur myélome (3,98 pour 100 patients-années) par rapport aux témoins (1,38 pour 100 patients-années). Les CS non invasifs sont essentiellement des épithéliomas basocellulaire ou spinocellulaire. La majorité des CS invasifs étaient des tumeurs malignes solides.
Dans les études cliniques menées chez des patients présentant un myélome multiple non préalablement traité non éligibles à une greffe, une incidence 4,9 fois plus élevée de CS hématologiques (cas de LAM et SMD) a été observée chez les patients recevant le lénalidomide en association avec le melphalan et la prednisone jusqu’à la progression de la maladie (1,75 pour 100 patients-années) par rapport aux patients recevant le melphalan en association avec la prednisone (0,36 pour 100 patients-années).
Une incidence 2,12 fois plus élevée de tumeurs solides secondaires a été observée chez les patients recevant le lénalidomide (9 cycles) en association avec le melphalan et la prednisone (1,57 pour 100 patients-années) par rapport aux patients recevant le melphalan en association avec la prednisone (0,74 pour 100 patients-années).
Chez les patients recevant le lénalidomide en association avec la dexaméthasone jusqu’à la progression de la maladie ou pendant 18 mois, l’incidence de CS hématologiques (0,16 pour 100 patients-années) n’a pas été plus élevé que chez les patients traités par le thalidomide en association avec le melphalan et la prednisone (0,79 pour 100 patients-années).
Une incidence 1,3 fois plus élevée de tumeurs solides secondaires a été observée chez les patients recevant le lénalidomide en association avec la dexaméthasone jusqu’à la progression de la maladie ou pendant 18 mois (1,58 pour 100 patients-années) par rapport aux patients recevant le thalidomide en association avec le melphalan et la prednisone (1,19 pour 100 patients-années).
Chez les patients atteints d’un myélome multiple non préalablement traité recevant le lénalidomide en association avec le bortézomib et la dexaméthasone, l’incidence de CS hématologiques était de 0,00 à 0,16 pour 100 patients-années et celle des tumeurs solides secondaires était de 0,21 à 1,04 pour 100 patients-années.
Le risque accru de cancers secondaires associé au lénalidomide est également observé dans le cas du MMNPT après une greffe de cellules souches. Bien que ce risque ne soit pas encore totalement caractérisé, il doit être pris en compte lorsqu’un traitement par Lénalidomide est envisagé et utilisé dans ce contexte.
L’incidence des cancers hématologiques, notamment les LAM, les SMD et les hémopathies malignes lymphoïdes B (dont le lymphome de Hodgkin), était de 1,31 pour 100 patients-années pour les bras lénalidomide et de 0,58 pour 100 patients-années pour les bras placebo (1,02 pour 100 patients-années pour les patients exposés au lénalidomide après une AGCS et 0,60 pour 100 patients-années pour les patients non exposés au lénalidomide après une AGCS). L’incidence des tumeurs solides secondaires était de 1,36 pour 100 patients-années pour les bras lénalidomide et de 1,05 pour 100 patients-années pour les bras placebo (1,26 pour 100 patients-années pour les patients exposés au lénalidomide après une AGCS et 0,60 pour 100 patients-années pour les patients non exposés au lénalidomide après une AGCS).
Le risque de survenue d’un cancer secondaire hématologique doit être pris en compte avant d’instaurer le traitement par le lénalidomide en association avec le melphalan ou immédiatement après le melphalan à forte dose et une autogreffe de cellules souches. Les médecins doivent évaluer soigneusement les patients avant et pendant le traitement en utilisant les méthodes habituelles de dépistage des cancers pour surveiller le développement de CS et instaurer un traitement s’il est indiqué.
Progression en leucémie aiguë myéloblastique dans les SMD à risque faible ou intermédiaire-1
· Caryotype
La présence d’anomalies cytogénétiques complexes au bilan initial est associée à une progression en LAM chez les patients dépendants transfusionnels et porteurs d’une anomalie Del (5q). Dans une analyse combinée de deux études cliniques du lénalidomide dans les syndromes myélodysplasiques à risque faible ou intermédiaire 1, le risque estimé cumulé à 2 ans de progression en LAM était le plus élevé chez les patients porteurs d’anomalies cytogénétiques complexes (38,6 %). Le taux estimé à 2 ans de progression en LAM chez les patients porteurs d’une anomalie Del (5q) isolée était de 13,8 % comparé à 17,3 % chez les patients porteurs de la délétion 5q et d’une autre anomalie cytogénétique.
Par conséquent, le rapport bénéfice/risque du lénalidomide lorsque le SMD est associé à la délétion 5q et à des anomalies cytogénétiques complexes est inconnu.
· Statut TP53
Une mutation du gène TP53 est présente chez 20 à 25 % des patients présentant un SMD Del (5q) à faible risque et est associée à un risque plus élevé de progression en leucémie aiguë myéloblastique (LAM). Dans une analyse post hoc d’une étude clinique du lénalidomide dans les syndromes myélodysplasiques à risque faible ou intermédiaire 1 (MDS-004), le taux estimé à 2 ans de progression en LAM était de 27,5 % chez les patients IHC-p53 positif et de 3,6 % chez les patients IHC-p53 négatif (p = 0,0038) (seuil du test de 1 % de forte coloration nucléaire, en utilisant l’analyse immunohistochimique de la protéine p53 comme marqueur de substitution du statut mutationnel de TP53) (voir rubrique 4.8).
Progression en d’autres cancers dans le lymphome à cellules du manteau
Dans le lymphome à cellules du manteau, la LAM, les hémopathies malignes lymphoïdes B et les cancers cutanés non mélanocytaires sont des risques identifiés.
Cancers secondaires au traitement dans le lymphome folliculaire
Dans une étude clinique portant sur les lymphomes non hodgkiniens indolents (LNHi) en rechute ou réfractaire dans laquelle des patients atteints d’un lymphome folliculaire ont été inclus, aucun risque accru de CS n’a été observé dans le bras lénalidomide/rituximab par rapport au bras placebo/rituximab. Les taux de CS hématologiques de type LAM étaient de 0,29 pour 100 patients-années versus 0,29 pour 100 patients-années chez les patients recevant le placebo/rituximab. L’incidence des tumeurs solides secondaires (à l’exclusion des cancers cutanés non mélanomateux) et des CS hématologiques était de 0,87 pour 100 patients-années dans le bras lénalidomide/rituximab versus 1,17 pour 100 patients-années chez les patients recevant le placebo/rituximab, avec un suivi médian de 30,59 mois (de 0,6 à 50,9 mois).
Il existe un risque identifié de cancer cutané non mélanomateux, notamment de type épithélioma spinocellulaire ou basocellulaire.
Les médecins doivent surveiller la survenue de CS chez les patients. Il convient de prendre en considération à la fois le bénéfice potentiel du lénalidomide et le risque de CS lorsqu’un traitement par le lénalidomide est envisagé.
Affections hépatiques
Une insuffisance hépatique, parfois d’issue fatale, a été rapportée chez des patients traités par le lénalidomide en association : des cas d’insuffisance hépatique aiguë, d’hépatite toxique, d’hépatite cytolytique, d’hépatite cholestatique et d’hépatite mixte cytolytique/cholestatique ont été rapportés. Les mécanismes de cette hépatotoxicité sévère d’origine médicamenteuse restent inconnus, même si, dans certains cas, une hépatopathie virale préexistante, des enzymes hépatiques élevées préalablement à la mise en œuvre du traitement et peut-être un traitement par des antibiotiques peuvent constituer des facteurs de risque.
Des anomalies des paramètres hépatiques ont fréquemment été rapportées et étaient généralement asymptomatiques et réversibles après l’interruption du traitement. Une fois les paramètres revenus aux valeurs initiales, la reprise du traitement à une dose plus faible peut être envisagée.
Le lénalidomide est éliminé par voie rénale. Il est important d’adapter la posologie chez les patients présentant une insuffisance rénale afin de rester en deçà des taux plasmatiques susceptibles de majorer l’hématotoxicité ou l’hépatotoxicité. Il est recommandé de surveiller la fonction hépatique, en particulier en cas d’antécédents ou de présence d’une infection hépatique d’origine virale ou lorsque le lénalidomide est associé à des médicaments connus pour induire une toxicité hépatique.
Infection avec ou sans neutropénie
Les patients atteints de myélome multiple ont tendance à développer des infections, dont des pneumonies. Il a été observé une fréquence plus élevée d’infections avec le lénalidomide en association avec la dexaméthasone qu’avec le MPT chez les patients atteints de myélome multiple non préalablement traité non éligibles à une greffe, de même qu’avec un traitement d’entretien par lénalidomide par rapport au placebo chez les patients atteints de myélome multiple non préalablement traité qui ont reçu une AGCS. Des infections de Grade ≥ 3 sont survenues dans le contexte d’une neutropénie chez moins d’un tiers des patients. Les patients présentant des facteurs de risque d’infection doivent être surveillés étroitement. Tous les patients doivent être informés qu’ils doivent consulter un médecin sans attendre au premier signe d’infection (par exemple toux, fièvre, etc.), ce qui permettra une prise en charge précoce pour en diminuer la sévérité.
Réactivation virale
Des cas de réactivation virale ont été rapportés chez des patients traités par le lénalidomide, dont des cas graves de réactivation du virus du zona ou du virus de l’hépatite B (VHB).
Certains cas de réactivation virale ont eu une issue fatale.
Dans certains des cas, la réactivation du virus du zona a entraîné un zona disséminé, une méningite zostérienne ou un zona ophtalmique nécessitant l’interruption temporaire ou l’arrêt définitif du traitement par le lénalidomide et l’instauration d’un traitement antiviral adéquat.
De rares cas de réactivation de l’hépatite B ont été rapportés chez des patients recevant le lénalidomide qui avaient des antécédents d’infection par le virus de l’hépatite B. Certains de ces cas ont évolué vers une insuffisance hépatique aiguë et conduit à l’arrêt du traitement par le lénalidomide et à l’instauration d’un traitement antiviral adéquat. La sérologie VHB doit être déterminée avant l’instauration du traitement par le lénalidomide. Chez les patients présentant un résultat positif au test de dépistage de l’infection par le VHB, une consultation chez un médecin spécialisé dans le traitement de l’hépatite B est recommandée. La prudence s’impose en cas d’administration du lénalidomide chez des patients ayant des antécédents d’infection par le VHB, y compris les patients positifs pour les anticorps anti-Hbc mais négatifs pour l’AgHbs. Ces patients doivent être étroitement surveillés pendant toute la durée du traitement afin de détecter des signes et symptômes d’infection active par le VHB.
Leucoencéphalopathie multifocale progressive
Des cas de leucoencéphalopathie multifocale progressive (LEMP), dont certains d’issue fatale, ont été rapportés avec le lénalidomide. Des cas de LEMP ont été rapportés de plusieurs mois à plusieurs années après avoir commencé un traitement par lénalidomide. Des cas ont été généralement rapportés chez les patients prenant simultanément de la dexaméthasone ou après un traitement antérieur avec d’autres chimiothérapies immunosuppressives.
Les médecins doivent surveiller les patients à intervalles réguliers et un diagnostic différentiel de LEMP doit être envisagé chez les patients présentant une aggravation ou de nouveaux symptômes neurologiques, signes ou symptômes cognitifs ou comportementaux. Il conviendra de conseiller aux patients d’informer leur conjoint ou le personnel soignant de leur traitement, ceux-ci pouvant remarquer des symptômes dont les patients ne sont pas conscients.
Le diagnostic d’une LEMP doit reposer sur un examen neurologique, une imagerie par résonance magnétique du cerveau et un dosage de l’ADN du virus JC (JCV) dans le liquide céphalo-rachidien par réaction en chaîne par polymérisation (PCR) ou une biopsie cérébrale suivie d’un test de dépistage du JCV. Une analyse négative par PCR ne permet pas d’écarter une LEMP. Une surveillance et des analyses complémentaires seront éventuellement justifiées si un diagnostic alternatif ne peut être établi.
Si une LEMP est suspectée, un dosage supplémentaire doit être suspendu jusqu’à ce que la LEMP soit exclue. Si la LEMP est confirmée, le lénalidomide doit être arrêté définitivement.
Patients atteints de myélome multiple non préalablement traité
La proportion d'événements indésirables (événements indésirables de Grade 3 ou 4, événements indésirables graves, arrêts du traitement) a été plus élevée chez les patients âgés de plus de 75 ans présentant une maladie de stade ISS (International staging system) III et ayant un indice de performance ECOG ≥ 2 ou une ClCr < 60 mL/min lorsque le lénalidomide était administré en association. La capacité des patients à tolérer le lénalidomide en association doit être évaluée attentivement en tenant compte de l'âge, le stade ISS III, l’indice de performance ECOG ≥ 2 ou la ClCr < 60 mL/min (voir rubriques 4.2 et 4.8).
Cataracte
Des cas de cataracte ont été rapportés à une fréquence plus élevée chez les patients recevant le lénalidomide en association avec la dexaméthasone, en particulier en cas de traitement au long cours. Des contrôles de l’acuité visuelle à intervalles réguliers sont recommandés.
Ce médicament contient moins de 1 mmol (23 mg) de sodium par gélule, c.-à-d. qu’il est essentiellement « sans sodium ».
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Contraceptifs oraux
Aucune étude d’interaction n’a été réalisée concernant les contraceptifs oraux.
Le lénalidomide n’est pas un inducteur enzymatique. Lors d’une étude in vitro sur des hépatocytes humains, le lénalidomide, aux diverses concentrations testées, n’a pas eu d’effet inducteur sur les enzymes CYP1A2, CYP2B6, CYP2C9, CYP2C19 et CYP3A4/5. Par conséquent, aucune induction entraînant une réduction d’efficacité des médicaments, notamment des contraceptifs hormonaux, n’est attendue en cas d’administration de lénalidomide seul. La dexaméthasone est néanmoins connue pour être un inducteur faible à modéré du CYP3A4 et risque probablement d’affecter d’autres enzymes ainsi que des protéines de transport. Il ne peut être exclu que l’efficacité des contraceptifs oraux se trouve réduite lors du traitement. Les mesures efficaces nécessaires doivent être prises pour éviter toute grossesse (voir rubriques 4.4 et 4.6).
Warfarine
L’administration concomitante de doses multiples de 10 mg de lénalidomide n’a eu aucun effet sur les caractéristiques pharmacocinétiques d’une dose unique de warfarine R- et S-. L’administration concomitante d’une dose unique de 25 mg de warfarine n’a eu aucun effet sur les caractéristiques pharmacocinétiques du lénalidomide. On ignore toutefois s’il existe des interactions dans le cadre d’une utilisation clinique (en association avec la dexaméthasone). La dexaméthasone est un inducteur enzymatique faible à modéré et ses effets sur la warfarine ne sont pas connus. Une surveillance étroite de la concentration de la warfarine est conseillée pendant le traitement.
Digoxine
L’administration concomitante de lénalidomide 10 mg en une prise par jour a accru l’exposition plasmatique à la digoxine (0,5 mg, dose unique) de 14 % avec un IC (intervalle de confiance) à 90 % [0,52 % – 28,2 %]. On ignore si les effets seraient différents en pratique clinique (doses du lénalidomide plus élevées et administration concomitante de dexaméthasone). Il est donc conseillé de surveiller la concentration de la digoxine pendant le traitement par le lénalidomide.
Statines
Il existe un risque accru de rhabdomyolyse lorsque les statines sont administrées avec le lénalidomide, qui peut être simplement additif. Une surveillance clinique et biologique renforcée est nécessaire, en particulier pendant les premières semaines de traitement.
Dexaméthasone
L’administration concomitante de doses uniques ou répétées de dexaméthasone (40 mg en une prise par jour) n’a pas eu d’effet cliniquement pertinent sur la pharmacocinétique à doses répétées du lénalidomide (25 mg en une prise par jour).
Interactions avec les inhibiteurs de la glycoprotéine P (P-gp)
In vitro, le lénalidomide est un substrat faible de la P-gp mais pas un inhibiteur de la P-gp. L’administration concomitante de doses répétées de quinidine, un inhibiteur puissant de la P-gp (600 mg deux fois par jour) ou de temsirolimus, un inhibiteur/substrat modéré de la P-gp (25 mg) n’a pas eu d’effet cliniquement pertinent sur la pharmacocinétique du lénalidomide (25 mg). L’administration concomitante de lénalidomide ne modifie pas la pharmacocinétique du temsirolimus.
4.6. Fertilité, grossesse et allaitement
Du fait de son potentiel tératogène, le lénalidomide doit être prescrit dans le cadre d’un Programme de Prévention de la Grossesse (voir rubrique 4.4), à moins de pouvoir affirmer avec certitude que la patiente est dans l’impossibilité de procréer.
Femmes en âge de procréer / Contraception chez les hommes et les femmes
Les femmes en âge de procréer doivent recourir à une méthode de contraception efficace. En cas de survenue d’une grossesse chez une femme traitée par le lénalidomide, le traitement doit être arrêté et la patiente doit être adressée à un médecin spécialiste ou expérimenté en tératologie pour évaluation et conseil. En cas de survenue d’une grossesse chez la partenaire d’un homme traité par le lénalidomide, il est recommandé d’adresser la femme à un médecin spécialiste ou expérimenté en tératologie pour évaluation et conseil.
Le lénalidomide est présent dans le sperme humain à des concentrations extrêmement faibles pendant le traitement et il est indétectable 3 jours après l’arrêt du médicament chez les sujets sains (voir rubrique 5.2). À titre de précaution, et en tenant compte de l’allongement du temps d’élimination dans les populations particulières telles que les patients présentant une insuffisance rénale, tous les hommes traités par le lénalidomide doivent donc utiliser des préservatifs pendant toute la durée du traitement, y compris en cas d’interruption des prises, et pendant 1 semaine à l’issue du traitement si leur partenaire est enceinte ou en âge de procréer et n’utilise pas de méthode contraceptive.
Grossesse
Le lénalidomide est structurellement proche du thalidomide. Le thalidomide est une substance active tératogène connue chez l’homme, provoquant des anomalies congénitales graves, potentiellement létales chez l’enfant à naître.
Le lénalidomide induit chez les singes des malformations similaires à celles décrites avec le thalidomide (voir rubrique 5.3). Par conséquent, un effet tératogène du lénalidomide est attendu et l’utilisation du lénalidomide au cours de la grossesse est contre-indiquée (voir rubrique 4.3).
Allaitement
On ignore si le lénalidomide est excrété dans le lait maternel. L’allaitement doit donc être interrompu pendant le traitement par le lénalidomide.
Fertilité
Dans une étude de fertilité chez le rat, des doses de lénalidomide allant jusqu’à 500 mg/kg (environ 200 à 500 fois respectivement les doses humaines de 25 mg et 10 mg sur la base de la surface corporelle) n’ont pas entraîné d’effets indésirables sur la fertilité, ni de toxicité parentale.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Des cas de fatigue, étourdissements, somnolence, vertiges et vision trouble ont été signalés lors de l’utilisation du lénalidomide. La conduite de véhicules ou l’utilisation de machines devront donc se faire avec précaution.
Résumé du profil de sécurité
· Myélome multiple non préalablement traité : patients recevant le lénalidomide en traitement d’entretien après une AGCS
Une approche conservatrice a été adoptée pour déterminer les effets indésirables survenus au cours de l’étude CALGB 100104. Les effets indésirables décrits dans le tableau 1 incluaient les événements rapportés après un traitement par MFD/AGCS, ainsi que les événements survenus au cours de la période de traitement d'entretien. Une deuxième analyse ayant identifié les événements survenus après le début du traitement d’entretien suggère que les fréquences décrites dans le tableau 1 peuvent être plus élevées que celles observées pendant la période de traitement d’entretien. Dans l'étude IFM 2005-02, les effets indésirables ne concernaient que la période de traitement d'entretien.
Les effets indésirables graves observés plus fréquemment (≥ 5 %) avec le lénalidomide en traitement d’entretien qu’avec le placebo ont été :
o Pneumonie (10,6 % ; terme combiné) dans l'étude IFM 2005-02
o Infection pulmonaire (9,4 % [9,4 % après le début du traitement d’entretien]) dans l'étude CALGB 100104
Dans l’étude IFM 2005-02, les effets indésirables observés plus fréquemment avec le lénalidomide en traitement d’entretien qu’avec le placebo ont été : neutropénie (60,8 %), bronchite (47,4 %), diarrhée (38,9 %), rhinopharyngite (34,8 %), spasmes musculaires (33,4 %), leucopénie (31,7 %), asthénie (29,7 %), toux (27,3 %), thrombopénie (23,5 %), gastro-entérite (22,5 %) et pyrexie (20,5 %).
Dans l’étude CALGB 100104, les effets indésirables observés plus fréquemment avec le lénalidomide en traitement d’entretien qu’avec le placebo ont été : neutropénie (79,0 % [71,9 % après le début du traitement d’entretien]), thrombopénie (72,3 % [61,6 %]), diarrhée (54,5 % [46,4 %]), éruption cutanée (31,7 % [25,0 %]), infection des voies respiratoires supérieures (26,8 % [26,8 %]), fatigue (22,8 % [17,9 %]), leucopénie (22,8 % [18,8 %]) et anémie (21,0 % [13,8 %]).
· Myélome multiple non préalablement traité : patients non éligibles à une greffe recevant le lénalidomide en association avec le bortézomib et la dexaméthasone
Dans l’étude SWOG S0777, les effets indésirables graves observés plus fréquemment (≥ 5 %) avec le lénalidomide en association avec le bortézomib intraveineux et la dexaméthasone qu’avec le lénalidomide en association avec la dexaméthasone ont été :
o Hypotension (6,5 %)
o Infection pulmonaire (5,7 %)
o Déshydratation (5,0 %)
Les effets indésirables observés plus fréquemment avec le lénalidomide en association avec le bortézomib et la dexaméthasone qu’avec le lénalidomide en association avec la dexaméthasone ont été : fatigue (73,7 %), neuropathie périphérique (71,8 %), thrombopénie (57,6 %), constipation (56,1 %), hypocalcémie (50,0 %).
· Myélome multiple non préalablement traité : patients non éligibles à une greffe, traités par le lénalidomide en association avec la dexaméthasone à faible dose
Les effets indésirables graves observés plus fréquemment (≥ 5 %) avec l’association lénalidomide + dexaméthasone à faible dose (Rd et Rd18) qu’avec l’association melphalan, prednisone et thalidomide (MPT) ont été :
o Pneumonie (9,8 %)
o Insuffisance rénale (y compris aiguë) (6,3 %)
Les effets indésirables observés plus fréquemment dans les bras Rd ou Rd18 que dans le bras MPT ont été : diarrhée (45,5 %), fatigue (32,8 %), dorsalgies (32,0 %), asthénie (28,2 %), insomnies (27,6 %), rash (24,3 %), diminution de l’appétit (23,1 %), toux (22,7 %), pyrexie (21,4 %) et spasmes musculaires (20,5 %).
· Myélome multiple non préalablement traité : patients non éligibles à une greffe, traités par le lénalidomide en association avec le melphalan et la prednisone
Les effets indésirables graves observés plus fréquemment (≥ 5 %) avec l’association melphalan, prednisone et lénalidomide suivie du lénalidomide en traitement d’entretien (MPR+R) ou melphalan, prednisone et lénalidomide suivie du placebo (MPR+p) qu'avec l’association melphalan, prednisone et placebo suivie du placebo (MPp+p) ont été :
o Neutropénie fébrile (6,0 %)
o Anémie (5,3 %)
Les effets indésirables observés plus fréquemment dans les bras MPR+R ou MPR+p que dans le bras MPp+p ont été : neutropénie (83,3 %), anémie (70,7 %), thrombopénie (70,0 %), leucopénie (38,8 %), constipation (34,0 %), diarrhée (33,3 %), éruptions cutanées (28,9 %), pyrexie (27,0 %), œdème périphérique (25,0 %), toux (24,0 %), diminution de l’appétit (23,7 %) et asthénie (22,0 %).
· Myélome multiple : patients ayant reçu au moins un traitement antérieur
Lors de deux essais de phase 3 contrôlés par placebo, 353 patients atteints de myélomes multiples ont été traités par l’association lénalidomide/dexaméthasone tandis que 351 ont reçu l’association placebo/dexaméthasone.
Les effets indésirables les plus graves observés plus fréquemment avec l’association lénalidomide/dexaméthasone qu’avec l’association placebo/dexaméthasone ont été :
o Des accidents thrombo-emboliques veineux (thromboses veineuses profondes, embolies pulmonaires) (voir rubrique 4.4)
o Des neutropénies de Grade 4 (voir rubrique 4.4)
Les effets indésirables observés plus fréquemment avec le lénalidomide + dexaméthasone qu’avec le placebo + dexaméthasone dans les essais cliniques combinés menés dans le myélome multiple (MM-009 et MM-010), ont été : fatigue (43,9 %), neutropénie (42,2 %), constipation (40,5 %), diarrhée (38,5 %), spasmes musculaires (33,4 %), anémie (31,4 %), thrombopénie (21,5 %) et éruptions cutanées (21,2 %).
· Syndromes myélodysplasiques
Le profil de sécurité global du lénalidomide chez les patients présentant un syndrome myélodysplasique est issu des données de 286 patients au total ayant participé à une étude de phase 2 et à une étude de phase 3 (voir la rubrique 5.1). Dans l’étude de phase 2, les 148 patients ont reçu le lénalidomide. Dans l’étude de phase 3, 69 patients ont été traités par le lénalidomide à 5 mg, 69 patients par le lénalidomide à 10 mg et 67 patients ont reçu du placebo dans la phase en double aveugle de l’étude.
La majorité des effets indésirables ont tendance à survenir au cours des 16 premières semaines de traitement par le lénalidomide.
Les effets indésirables graves ont été :
o Des accidents thrombo-emboliques veineux (thrombose veineuse profonde, embolie pulmonaire) (voir rubrique 4.4)
o Des neutropénies de Grade 3 ou 4, neutropénies fébriles et thrombopénies de Grade 3 ou 4 (voir rubrique 4.4)
Dans l’étude de phase 3, les effets indésirables observés le plus fréquemment dans les groupes traités par le lénalidomide par rapport au groupe contrôle ont été : la neutropénie (76,8 %), la thrombopénie (46,4 %), la diarrhée (34,8 %), la constipation (19,6 %), les nausées (19,6 %), le prurit (25,4 %), l’éruption cutanée (18,1 %), la fatigue (18,1 %) et les spasmes musculaires (16,7 %).
· Lymphome à cellules du manteau
Le profil de sécurité global du lénalidomide chez les patients présentant un lymphome à cellules du manteau est issu des données de 254 patients ayant participé à l’étude de phase 2 randomisée contrôlée MCL-002 (voir rubrique 5.1).
De plus, les effets indésirables rapportés dans l’étude de confirmation MCL-001 ont été inclus dans le tableau 3.
Les effets indésirables graves observés plus fréquemment (avec une différence d’au moins 2 %) dans le bras traité par le lénalidomide que dans le bras contrôle dans l’étude MCL-002 ont été :
o Neutropénie (3,6 %)
o Embolie pulmonaire (3,6 %)
o Diarrhée (3,6 %)
Les effets indésirables les plus fréquents survenus plus fréquemment dans le bras traité par le lénalidomide que dans le bras contrôle dans l’étude MCL-002 ont été : neutropénie (50,9 %), anémie (28,7 %), diarrhée (22,8 %), fatigue (21,0 %), constipation (17,4 %), pyrexie (16,8 %) et éruption cutanée (y compris dermatite allergique) (16,2 %).
Dans l’étude MCL-002, il a été globalement observé une augmentation apparente des décès prématurés (au cours des 20 premières semaines). Les patients ayant une charge tumorale élevée avant l’initiation du traitement présentent un risque accru de décès prématuré : des décès prématurés ont été rapportés chez 16/81 patients (20 %) du bras lénalidomide et 2/28 patients (7 %) du bras contrôle. Les chiffres correspondants sur 52 semaines étaient de 32/81 patients (39,5 %) et 6/28 patients (21 %) (voir rubrique 5.1).
Pendant le premier cycle de traitement, le traitement a été arrêté chez 11/81 patients (14 %) ayant une charge tumorale élevée du bras lénalidomide versus 1/28 patients (4 %) du bras contrôle. La survenue d’événements indésirables était la principale raison d’arrêt du traitement pendant le cycle 1 chez les patients ayant une charge tumorale élevée (7/11, 64 %). Une charge tumorale élevée était définie comme la présence d’au moins une lésion mesurant ≥ 5 cm de diamètre ou d’au moins 3 lésions mesurant ≥ 3 cm.
· Lymphome folliculaire
Le profil de sécurité global du lénalidomide en association avec le rituximab chez les patients présentant un lymphome folliculaire, préalablement traité est issu des données de 294 patients ayant participé à l’étude de phase 3 randomisée contrôlée NHL-007. De plus, les effets indésirables rapportés dans l’étude de confirmation NHL-008 ont été inclus dans le tableau 5.
Les effets indésirables graves observés plus fréquemment (avec une différence d’au moins 1 %) dans le bras traité par le lénalidomide/rituximab que dans le bras placebo/rituximab dans l’étude NHL-007 ont été :
o Neutropénie fébrile (2,7 %)
o Embolie pulmonaire (2,7 %)
o Pneumonie (2,7 %)
Les effets indésirables survenus plus fréquemment dans le bras traité par le lénalidomide/rituximab que dans le bras placebo/rituximab dans l’étude NHL-007 (avec une fréquence plus élevée d’au moins 2 %) ont été : neutropénie (58,2 %), diarrhée (30,8 %), leucopénie (28,8 %), constipation (21,9 %), toux (21,9 %) et fatigue (21,9 %).
Liste tabulée des effets indésirables
Les effets indésirables observés chez les patients traités par le lénalidomide sont présentés ci-dessous par classe de systèmes d’organes et fréquence. Au sein de chaque groupe de fréquence, les effets indésirables sont présentés suivant un ordre décroissant de gravité. Les fréquences sont définies comme suit : très fréquent (≥ 1/10) ; fréquent (≥ 1/100 à < 1/10) ; peu fréquent (≥ 1/1 000 à < 1/100) ; rare (≥ 1/10 000 à < 1/1 000) ; très rare (< 1/10 000) ; fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
Les effets indésirables ont été inclus dans la catégorie appropriée dans le tableau ci-dessous en fonction de la fréquence la plus élevée observée dans l’une des études cliniques principales.
Résumé tabulé pour le traitement en monothérapie dans le myélome multiple
Le tableau suivant résulte des données collectées lors des études menées chez des patients atteints de myélome multiple non préalablement traité recevant le lénalidomide en traitement d’entretien après une AGCS. Les données n’ont pas été corrigées en fonction de la durée de traitement plus longue dans les bras de traitement par lénalidomide poursuivi jusqu’à progression de la maladie par rapport aux bras placebo dans les études pivots menées dans le myélome multiple (voir rubrique 5.1).
Tableau 1. Effets indésirables rapportés lors des études cliniques chez les patients atteints de myélome multiple recevant le lénalidomide en traitement d’entretien
|
Classe de systèmes d’organes / Terme préférentiel |
Effets indésirables de tous Grades / Fréquence |
Effets indésirables de Grade 3 − 4 / Fréquence |
|
Infections et infestations |
Très fréquent Pneumonie◊,a, infection des voies respiratoires supérieures, infection neutropénique, bronchite◊, grippe◊, gastro-entérite◊, sinusite, rhinopharyngite, rhinite. Fréquent Infection◊, infection urinaire◊,*, infection des voies respiratoires inférieures, infection pulmonaire◊ |
Très fréquent Pneumonie◊,a, infection neutropénique Fréquent Septicémie◊,b, bactériémie, infection pulmonaire◊, infection bactérienne des voies respiratoires inférieures, bronchite◊, grippe◊, gastro-entérite◊, zona◊, infection◊ |
|
Tumeurs bénignes, malignes et non précisées (incluant kystes et polypes) |
Fréquent Syndrome myélodysplasique◊,* |
|
|
Affections hématologiques et du système lymphatique |
Très fréquent Neutropénie^,◊, neutropénie fébrile^,◊, thrombopénie^,◊, anémie, leucopénie◊, lymphopénie |
Très fréquent Neutropénie^,◊, neutropénie fébrile^,◊,thrombopénie^,◊, anémie, leucopénie◊, lymphopénie Fréquent Pancytopénie◊ |
|
Troubles du métabolisme et de la nutrition |
Très fréquent Hypokaliémie |
Fréquent Hypokaliémie, déshydratation |
|
Affections du système nerveux |
Très fréquent Paresthésie Fréquent Neuropathie périphériquec |
Fréquent Céphalées |
|
Affections vasculaires |
Fréquent Embolie pulmonaire◊,* |
Fréquent Thrombose veineuse profonde^,◊,d |
|
Affections respiratoires, thoraciques et médiastinales |
Très fréquent Toux Fréquent Dyspnée◊, rhinorrhée |
Fréquent Dyspnée◊ |
|
Affections gastro-intestinales |
Très fréquent Diarrhée, constipation, douleur abdominale, nausées Fréquent Vomissements, douleur abdominale haute |
Fréquent Diarrhée, vomissements, nausées |
|
Affections hépatobiliaires |
Très fréquent Perturbation du bilan hépatique |
Fréquent Perturbation du bilan hépatique |
|
Affections de la peau et du tissu sous-cutané |
Très fréquent Éruption cutanée, sécheresse cutanée |
Fréquent Éruption cutanée, prurit |
|
Affections musculosquelettiques et systémiques |
Très fréquent Spasmes musculaires Fréquent Myalgies, douleur musculosquelettique |
|
|
Troubles généraux et anomalies au site d'administration |
Très fréquent Fatigue, asthénie, pyrexie |
Fréquent Fatigue, asthénie |
◊ Effets indésirables rapportés comme graves dans les études cliniques menées chez des patients présentant un myélome multiple non préalablement traité ayant reçu une AGCS.
* S'applique uniquement aux effets indésirables graves.
^ Voir rubrique 4.8 Description de certains effets indésirables.
a Le terme combiné « pneumonie » inclut les TP suivants : bronchopneumonie, pneumonie lobaire, pneumonie à Pneumocystis jiroveci, pneumonie, pneumonie à Klebsiella, pneumonie à Legionella, pneumonie à mycoplasme, pneumonie à pneumocoque, pneumonie à streptocoque, pneumonie virale, affection pulmonaire, pneumopathie.
b Le terme combiné « sepsis » inclut les TP suivants : sepsis bactérien, sepsis à pneumocoque, sepsis, choc septique, sepsis à staphylocoque.
c Le terme combiné « neuropathie périphérique » inclut les termes préférentiels (TP) suivants : neuropathie périphérique, neuropathie périphérique sensitive, polyneuropathie.
d Le terme combiné « thrombose veineuse profonde » inclut les TP suivants : thrombose veineuse profonde, thrombose, thrombose veineuse.
Résumé tabulé pour le traitement en association dans le myélome multiple
Le tableau suivant résulte des données collectées lors des études menées dans le myélome multiple avec le traitement en association. Les données n’ont pas été corrigées en fonction de la durée de traitement plus longue dans les bras de traitement par lénalidomide poursuivi jusqu’à progression de la maladie par rapport aux bras contrôles dans les études pivots menées dans le myélome multiple (voir rubrique 5.1).
Tableau 2. Effets indésirables rapportés lors des études cliniques chez les patients atteints de myélome multiple traités par le lénalidomide en association avec le bortézomib et la dexaméthasone, la dexaméthasone ou avec le melphalan et la prednisone
|
Classe de systèmes d’organes / Terme préférentiel |
Effets indésirables de tous Grades / Fréquence |
Effets indésirables de Grade 3 − 4 / Fréquence |
|
Infections et infestations |
Très fréquent Pneumonie◊,◊◊, infection des voies respiratoires supérieures◊, infections bactériennes, virales et fongiques (y compris infections opportunistes)◊, rhinopharyngite, pharyngite, bronchite◊, rhinite Fréquent Septicémie◊,◊◊, infection pulmonaire◊◊, infection des voies urinaires◊◊, sinusite◊ |
Fréquent Pneumonie◊,◊◊, infections bactériennes, virales et fongiques (y compris infections opportunistes)◊, cellulite◊, sepsis◊,◊◊, infection pulmonaire◊◊, bronchite◊, infection des voies respiratoires◊◊, infection des voies urinaires◊◊, entérocolite infectieuse |
|
Tumeurs bénignes, malignes et non précisées (incluant kystes et polypes) |
Peu fréquent Épithélioma basocellulaire^,◊, épithélioma spinocellulaire^,◊,* |
Fréquent Leucémie aiguë myéloblastique◊, syndrome myélodysplasique◊, épithélioma spinocellulaire^,◊,** Peu fréquent Leucémie aiguë à cellules T◊, épithélioma basocellulaire^,◊, syndrome de lyse tumorale |
|
Affections hématologiques et du système lymphatique |
Très fréquent Neutropénie^,◊,◊◊, thrombopénie^,◊,◊◊, anémie◊, troubles hémorragiques^, leucopénie, lymphopénie Fréquent Neutropénie fébrile^,◊, pancytopénie◊ Peu fréquent Hémolyse, anémie hémolytique auto-immune, anémie hémolytique |
Très fréquent Neutropénie^,◊,◊◊, thrombopénie^,◊,◊◊, anémie◊, leucopénie, lymphopénie Fréquent Neutropénie fébrile^,◊, pancytopénie◊, anémie hémolytique Peu fréquent Hypercoagulation, coagulopathie |
|
Affections du système immunitaire |
Peu fréquent Hypersensibilité^ |
|
|
Affections endocriniennes |
Fréquent Hypothyroïdie |
|
|
Troubles du métabolisme et de la nutrition |
Très fréquent Hypokaliémie◊,◊◊, hyperglycémie, hypoglycémie, hypocalcémie◊, hyponatrémie◊, déshydratation◊◊, diminution de l’appétit◊◊, perte de poids Fréquent Hypomagnésémie, hyperuricémie, hypercalcémie+ |
Fréquent Hypokaliémie◊,◊◊, hyperglycémie, hypocalcémie◊, diabète◊, hypophosphatémie, hyponatrémie◊, hyperuricémie, goutte, déshydratation◊◊, diminution de l’appétit◊◊, perte de poids |
|
Affections psychiatriques |
Très fréquent Dépression, insomnie Peu fréquent Perte de libido |
Fréquent Dépression, insomnie |
|
Affections du système nerveux |
Très fréquent Neuropathies périphériques◊◊, paresthésies, vertiges◊◊, tremblements, dysgueusie, céphalées Fréquent Ataxie, troubles de l’équilibre, syncope◊◊, névralgies, dysesthésie |
Très fréquent Neuropathies périphériques◊◊ Fréquent Accident vasculaire cérébral◊, vertiges◊◊, syncope◊◊, névralgies Peu fréquent Hémorragie intracrânienne^, accident ischémique transitoire, ischémie cérébrale |
|
Affections oculaires |
Très fréquent Cataracte, vision trouble Fréquent Diminution de l’acuité visuelle |
Fréquent Cataracte Peu fréquent Cécité |
|
Affections de l’oreille et du labyrinthe |
Fréquent Surdité (y compris hypoacousie), acouphènes |
|
|
Affections cardiaques |
Fréquent Fibrillation auriculaire◊,◊◊, bradycardie Peu fréquent Arythmie, allongement de l’intervalle QT, flutter auriculaire, extrasystoles ventriculaires |
Fréquent Infarctus du myocarde (y compris aigu)^,◊, fibrillation auriculaire◊,◊◊, insuffisance cardiaque congestive◊, tachycardie, insuffisance cardiaque◊,◊◊, ischémie myocardique◊ |
|
Affections vasculaires |
Très fréquent Événements thrombo-emboliques veineux^, essentiellement thrombose veineuse profonde et embolie pulmonaire^,◊,◊◊, hypotension◊◊ Fréquent Hypertension, ecchymoses^ |
Très fréquent Événements thrombo-emboliques veineux^, essentiellement thrombose veineuse profonde et embolie pulmonaire^,◊,◊◊ Fréquent Vascularite, hypotension◊◊, hypertension Peu fréquent Ischémie, ischémie périphérique, thrombose du sinus veineux intracrânien |
|
Affections respiratoires, thoraciques et médiastinales |
Très fréquent Dyspnée◊,◊◊, épistaxis^, toux Fréquent Dysphonie |
Fréquent Détresse respiratoire◊, dyspnée◊,◊◊, douleur pleurale◊◊, hypoxie◊◊ |
|
Affections gastro-intestinales |
Très fréquent Diarrhée◊,◊◊, constipation◊, douleur abdominale◊◊, nausées, vomissements◊◊, dyspepsie, sécheresse buccale, stomatite Fréquent Hémorragie gastro-intestinale (y compris hémorragie rectale, hémorragie hémorroïdaire, ulcère gastro-duodénal hémorragique et saignement gingival)^,◊◊, dysphagie Peu fréquent Colite, inflammation du caecum |
Fréquent Hémorragie gastro-intestinale^,◊,◊◊, occlusion de l’intestin grêle◊◊, diarrhée◊◊, constipation◊, douleur abdominale◊◊, nausées, vomissements◊◊ |
|
Affections hépatobiliaires |
Très fréquent Augmentation de l’alanine aminotransférase, augmentation de l’aspartate aminotransférase Fréquent Lésion hépatocellulaire◊◊, perturbation du bilan hépatique◊, hyperbilirubinémie Peu fréquent Insuffisance hépatique^ |
Fréquent Cholestase◊, hépatotoxicité, lésion hépatocellulaire◊◊, augmentation de l’alanine aminotransférase, perturbation du bilan hépatique◊ Peu fréquent Insuffisance hépatique^ |
|
Affections de la peau et du tissu sous cutané |
Très fréquent Éruptions cutanées◊◊, prurit Fréquent Urticaire, hyperhidrose, sécheresse cutanée, hyperpigmentation de la peau, eczéma, érythème Peu fréquent Réaction médicamenteuse accompagnée d’une éosinophilie et de symptômes systémiques (DRESS)◊◊, anomalie de la coloration cutanée, réaction de photosensibilité |
Fréquent Éruptions cutanées◊◊
Peu fréquent Réaction médicamenteuse accompagnée d’une éosinophilie et de symptômes systémiques (DRESS) ◊◊ |
|
Affections musculosquelettiques et systémiques |
Très fréquent Faiblesse musculaire◊◊, spasmes musculaires, douleur osseuse◊, douleur et gêne musculo-squelettique et du tissu conjonctif (y compris dorsalgies◊,◊◊), douleur dans les extrémités, myalgies, arthralgies◊ Fréquent Gonflement articulaire |
Fréquent Faiblesse musculaire◊◊, douleur osseuse◊, douleur et gêne musculo-squelettique et du tissu conjonctif (y compris dorsalgies◊,◊◊) Peu fréquent Gonflement articulaire |
|
Affections du rein et des voies urinaires |
Très fréquent Insuffisance rénale (y compris aiguë)◊,◊◊ Fréquent Hématurie^, rétention urinaire, incontinence urinaire Peu fréquent Syndrome de Fanconi acquis |
Peu fréquent Nécrose tubulaire rénale |
|
Affections des organes de reproduction et du sein |
Fréquent Dysfonctionnement érectile |
|
|
Troubles généraux et anomalies au site d’administration |
Très fréquent Fatigue◊,◊◊, œdème (y compris œdème périphérique), pyrexie◊,◊◊, asthénie, syndrome pseudo-grippal (y compris pyrexie, toux, myalgie, douleur musculo-squelettique, céphalées et raideurs) Fréquent Douleur thoracique◊,◊◊, léthargie |
Très fréquent Fatigue◊,◊◊ Fréquent Œdème périphérique, pyrexie◊,◊◊, asthénie |
|
Investigations |
Très fréquent Augmentation des phosphatases alcalines sanguines Fréquent Augmentation de la protéine C réactive |
|
|
Lésions, intoxications et complications liées aux procédures |
Fréquent Chute, contusion^ |
◊◊ Effets indésirables rapportés comme graves dans les études cliniques menées chez des patients présentant un myélome multiple non préalablement traité qui recevaient le lénalidomide en association avec le bortézomib et la dexaméthasone.
^ Voir rubrique 4.8 Description de certains effets indésirables.
◊ Effets indésirables rapportés comme graves dans les études cliniques menées chez des patients présentant un myélome multiple traités par le lénalidomide en association avec la dexaméthasone ou avec le melphalan et la prednisone.
+ S'applique uniquement aux effets indésirables graves.
* Des épithéliomas spinocellulaires ont été rapportés dans les études cliniques chez les patients atteints de myélome multiple préalablement traité recevant le lénalidomide en association avec la dexaméthasone par rapport aux témoins.
** Des épithéliomas spinocellulaires ont été rapportés dans une étude clinique chez des patients présentant un myélome multiple non préalablement traité recevant le lénalidomide en association avec la dexaméthasone par rapport aux témoins.
Résumé tabulé pour le traitement en monothérapie
Les tableaux suivants résultent des données collectées lors des principales études du lénalidomide en monothérapie dans les syndromes myélodysplasiques et le lymphome à cellules du manteau.
Tableau 3. Effets indésirables rapportés lors des études cliniques chez les patients présentant des syndromes myélodysplasiques traités par le lénalidomide#
|
Classe de systèmes d’organes / Terme préférentiel |
Effets indésirables de tous Grades/ Fréquence |
Effets indésirables de Grade 3 – 4 / Fréquence |
|
Infections et infestations |
Très fréquent Infections bactériennes, virales et fongiques (y compris infections opportunistes)◊ |
Très fréquent Pneumonie◊ Fréquent Infections bactériennes, virales et fongiques (y compris infections opportunistes)◊, bronchite |
|
Affections hématologiques et du système lymphatique |
Très fréquent Thrombopénie^,◊, neutropénie^,◊, anémie◊, leucopénie |
Très fréquent Thrombopénie^,◊, neutropénie^,◊, anémie◊, leucopénie Fréquent Neutropénie fébrile^,◊ |
|
Affections endocriniennes |
Très fréquent Hypothyroïdie |
|
|
Troubles du métabolisme et de la nutrition |
Très fréquent Diminution de l’appétit Fréquent Surcharge martiale, perte de poids |
Fréquent Hyperglycémie◊, diminution de l’appétit |
|
Affections psychiatriques |
Fréquent Troubles de l’humeur◊,~ |
|
|
Affections du système nerveux |
Très fréquent Étourdissements, céphalées Fréquent Paresthésies |
|
|
Affections cardiaques |
Fréquent Infarctus du myocarde aigu^,◊, fibrillation auriculaire◊, insuffisance cardiaque◊ |
|
|
Affections vasculaires |
Fréquent Hypertension, hématomes |
Fréquent Événements thrombo-emboliques veineux, essentiellement thrombose veineuse profonde et embolie pulmonaire^,◊ |
|
Affections respiratoires, thoraciques et médiastinales |
Très fréquent Épistaxis^ |
|
|
Affections gastro-intestinales |
Très fréquent Diarrhée◊, douleur abdominale (y compris douleur abdominale haute), nausées, vomissements, constipation Fréquent Sécheresse de la bouche, dyspepsie |
Fréquent Diarrhée◊, nausées, odontalgie |
|
Affections hépatobiliaires |
Fréquent Perturbation du bilan hépatique |
Fréquent Perturbation du bilan hépatique |
|
Affections de la peau et du tissu sous-cutané |
Très fréquent Éruptions cutanées, sécheresse cutanée, prurit |
Fréquent Éruptions cutanées, prurit |
|
Affections musculosquelettiques et systémiques |
Très fréquent Spasmes musculaires, douleur musculo-squelettique (incluant dorsalgies◊ et douleurs dans les membres), arthralgies, myalgies |
Fréquent Dorsalgies◊ |
|
Affections du rein et des voies urinaires |
Fréquent Insuffisance rénale◊ |
|
|
Troubles généraux et anomalies au site d’administration |
Très fréquent Fatigue, œdème périphérique, syndrome pseudo-grippal (y compris pyrexie, toux, pharyngite, myalgie, douleur musculo-squelettique, céphalées) |
Fréquent Pyrexie |
|
Lésions, intoxications et complications liées aux procédures |
Fréquent Chute |
^ Voir rubrique 4.8 Description de certains effets indésirables.
◊ Effets indésirables rapportés comme graves dans les études cliniques menées dans les syndromes myélodysplasiques.
~ Les troubles de l’humeur ont été un événement indésirable grave fréquent dans l’étude de phase 3 dans les syndromes myélodysplasiques ; aucun événement de Grade 3 ou 4 n’a été rapporté.
Algorithme utilisé pour l’inclusion dans le RCP : tous les effets indésirables enregistrés à l’aide de l’algorithme de l’étude de phase 3 sont répertoriés dans le RCP européen. Pour ces effets indésirables, un contrôle supplémentaire de la fréquence des effets indésirables enregistrés à l’aide de l’algorithme de l’étude de phase 2 a été effectué ; si la fréquence de l’effet indésirable dans l’étude de phase 2 était supérieure à celle rapportée dans l’étude de phase 3, l’événement a été répertorié dans le RCP européen dans la catégorie de fréquence observée dans l’étude de phase 2.
# Algorithme utilisé pour les syndromes myélodysplasiques :
· Étude de phase 3 dans les syndromes myélodysplasiques (population étudiée en tolérance en double aveugle, différence entre les groupes lénalidomide 5/10 mg et placebo, en fonction du schéma posologique initial, survenus chez au moins 2 patients)
o Tous les effets indésirables apparus sous traitement rapportés chez ≥ 5 % des patients traités par le lénalidomide et avec une différence d’au moins 2 % entre le lénalidomide et le placebo
o Tous les effets indésirables apparus sous traitement de Grade 3 ou 4 rapportés chez 1 % des patients traités par le lénalidomide et avec une différence d’au moins 1 % entre le lénalidomide et le placebo
o Tous les effets indésirables graves apparus sous traitement rapportés chez 1 % des patients traités par le lénalidomide et avec une différence d’au moins 1 % entre le lénalidomide et le placebo
· Étude de phase 2 dans les syndromes myélodysplasiques
o Tous les effets indésirables apparus sous traitement rapportés chez ≥ 5 % des patients traités par le lénalidomide
o Tous les effets indésirables apparus sous traitement de Grade 3 ou 4 rapportés chez 1 % des patients traités par le lénalidomide
o Tous les effets indésirables graves apparus sous traitement rapportés chez 1 % des patients traités par le lénalidomide
Tableau 4. Effets indésirables rapportés lors des études cliniques chez les patients présentant un lymphome à cellules du manteau traités par le lénalidomide
|
Classe de systèmes d’organes / Terme préférentiel |
Effets indésirables de tous Grades / Fréquence |
Effets indésirables de Grade 3 – 4/ Fréquence |
|
Infections et infestations |
Très fréquent Infections bactériennes, virales et fongiques (y compris infections opportunistes)◊, rhinopharyngite, pneumonie◊ Fréquent Sinusite |
Fréquent Infections bactériennes, virales et fongiques (y compris infections opportunistes)◊, pneumonie◊ |
|
Tumeurs bénignes, malignes et non précisées (incluant kystes et polypes) |
Fréquent Réaction de poussée tumorale |
Fréquent Réaction de poussée tumorale, épithélioma spinocellulaire^ ,◊, épithélioma basocellulaire^,◊ |
|
Affections hématologiques et du système lymphatique |
Très fréquent Thrombopénie^, neutropénie^,◊, leucopénie◊, anémie◊ Fréquent Neutropénie fébrile^,◊ |
Très fréquent Thrombopénie^, neutropénie^,◊, anémie◊ Fréquent Neutropénie fébrile^,◊, leucopénie◊ |
|
Troubles du métabolisme et de la nutrition |
Très fréquent Diminution de l’appétit, perte de poids, hypokaliémie Fréquent Déshydratation◊ |
Fréquent Déshydratation◊, hyponatrémie, hypocalcémie |
|
Affections psychiatriques |
Fréquent Insomnie |
|
|
Affections du système nerveux |
Très fréquent Dysgueusie, céphalées, neuropathie périphérique |
Fréquent Neuropathie sensitive périphérique, léthargie |
|
Affections de l’oreille et du labyrinthe |
Fréquent Vertiges |
|
|
Affections cardiaques |
Fréquent Infarctus du myocarde (y compris aigu)^,◊, insuffisance cardiaque |
|
|
Affections vasculaires |
Fréquent Hypotension◊ |
Fréquent Thrombose veineuse profonde◊, embolie pulmonaire^,◊, hypotension◊ |
|
Affections respiratoires, thoraciques et médiastinales |
Très fréquent Dyspnée◊ |
Fréquent Dyspnée◊ |
|
Affections gastro-intestinales |
Très fréquent Diarrhée◊, nausées◊, vomissements◊, constipation Fréquent Douleur abdominale |
Fréquent Diarrhée◊, douleur abdominale◊, constipation |
|
Affections de la peau et du tissu sous-cutané |
Très fréquent Éruptions cutanées (y compris dermatite allergique), prurit Fréquent Sueurs nocturnes, sécheresse cutanée |
Fréquent Éruptions cutanées |
|
Affections musculo-squelettiques et systémiques |
Très fréquent Spasmes musculaires, dorsalgies Fréquent Arthralgies, douleurs dans les extrémités, faiblesse musculaire◊ |
Fréquent Dorsalgies, faiblesse musculaire◊, arthralgies, douleurs dans les extrémités |
|
Affections du rein et des voies urinaires |
Fréquent Insuffisance rénale◊ |
|
|
Troubles généraux et anomalies au site d’administration |
Très fréquent Fatigue, asthénie◊, œdème périphérique, syndrome pseudo-grippal (y compris pyrexie◊, toux) Fréquent Frissons |
Fréquent Pyrexie◊, asthénie◊, fatigue |
^ Voir rubrique 4.8 Description de certains effets indésirables.
◊ Effets indésirables rapportés comme graves dans les études cliniques menées dans le lymphome à cellules du manteau. Algorithme utilisé pour le lymphome à cellules du manteau :
· Étude de phase 2 contrôlée dans le lymphome à cellules du manteau :
o Tous les effets indésirables apparus sous traitement rapportés chez ≥ 5 % des patients du bras lénalidomide et avec une différence d’au moins 2 % entre le bras lénalidomide et le bras contrôle
o Tous les effets indésirables apparus sous traitement de Grade 3 ou 4 rapportés chez ≥ 1 % des patients du bras lénalidomide et avec une différence d’au moins 1 % entre le bras lénalidomide et le bras contrôle
o Tous les effets indésirables graves apparus sous traitement rapportés chez ≥ 1 % des patients du bras lénalidomide et avec une différence d’au moins 1 % entre le bras lénalidomide et le bras contrôle
· Étude de phase 2 en un seul bras dans le lymphome à cellules du manteau :
o Tous les effets indésirables apparus sous traitement rapportés chez ≥ 5 % des patients
o Tous les effets indésirables apparus sous traitement de Grade 3 ou 4 rapportés chez au moins 2 patients
o Tous les effets indésirables graves apparus sous traitement rapportés chez au moins 2 patients
Résumé tabulé pour le traitement en association dans le lymphome folliculaire
Le tableau suivant résulte des données collectées lors des principales études (NHL-007 et NHL-008) du lénalidomide en association avec le rituximab menées chez des patients atteints d’un lymphome folliculaire.
Tableau 5. Effets indésirables rapportés lors des études cliniques chez les patients atteints d’un lymphome folliculaire, traités par le lénalidomide en association avec le rituximab
|
Classe de systèmes d’organes / Terme préférentiel |
Effets indésirables de tous Grades / Fréquence |
Effets indésirables de Grade 3 – 4 / Fréquence |
|
Infections et infestations |
Très fréquent Infection des voies respiratoires supérieures Fréquent Pneumonie◊, grippe, bronchite, sinusite, infection urinaire |
Fréquent Pneumonie◊, septicémie◊, infection pulmonaire, bronchite, gastro-entérite, sinusite, infection urinaire, cellulite◊ |
|
Tumeurs bénignes, malignes et non précisées (incluant kystes et polypes) |
Très fréquent Poussée tumorale^ Fréquent Epithélioma spinocellulaire◊,^,+ |
Fréquent Epithélioma basocellulaire^,◊ |
|
Affections hématologiques et du système lymphatiques |
Très fréquent Neutropénie^,◊, anémie◊, thrombopénie^, leucopénie**, lymphopénie*** |
Très fréquent Neutropénie^,◊ Fréquent Anémie◊, thrombopénie^, neutropénie fébrile◊, pancytopénie, leucopénie**, lymphopénie*** |
|
Troubles du métabolisme et de la nutrition |
Très fréquent Diminution de l’appétit, hypokaliémie Fréquent Hypophosphatémie, déshydratation |
Fréquent Déshydratation, hypercalcémie◊, hypokaliémie, hypophosphatémie, hyperuricémie |
|
Affections psychiatriques |
Fréquent Dépression, insomnie |
|
|
Affections du système nerveux |
Très fréquent Céphalées, vertiges Fréquent Neuropathie périphérique, dysgueusie |
Fréquent Syncope |
|
Affections cardiaques |
Peu fréquent Arythmie◊ |
|
|
Affections vasculaires |
Fréquent Hypotension |
Fréquent Embolie pulmonaire^,◊, hypotension |
|
Affections respiratoires, thoraciques et médiastinales |
Très fréquent Dyspnée◊, toux Fréquent Douleur oropharyngée, dysphonie |
Fréquent Dyspnée◊ |
|
Affections gastro-intestinales |
Très fréquent Douleur abdominale◊, diarrhée, constipation, nausées, vomissements, dyspepsie Fréquent Douleur abdominale haute, stomatite, sécheresse buccale |
Fréquent Douleur abdominale◊, diarrhée, constipation, stomatite |
|
Affection de la peau et du tissu sous-cutané |
Très fréquent Éruption cutanée*, prurit Fréquent Sécheresse cutanée, sueurs nocturnes, érythème |
Fréquent Éruption cutanée*, prurit |
|
Affections musculo-squelettiques et systémiques |
Très fréquent Spasmes musculaires, dorsalgies, arthralgies Fréquent Douleurs dans les extrémités, faiblesse musculaire, douleurs musculo-squelettiques, myalgies, cervicalgie |
Fréquent Faiblesse musculaire, cervicalgie |
|
Affections du rein et des voies urinaires |
|
Fréquent Atteinte rénale aiguë◊ |
|
Troubles généraux et anomalies au site d’administration |
Très fréquent Pyrexie, fatigue, asthénie, œdème périphérique Fréquent Malaise, frissons |
Fréquent Fatigue, asthénie |
|
Investigations |
Très fréquent Augmentation de l’alanine aminotransférase Fréquent Perte de poids, hyperbilirubinémie |
|
^ Voir rubrique 4.8 Description de certains effets indésirables.
Algorithme utilisé pour le lymphome folliculaire :
Étude de phase 3 – contrôlée :
o NHL-007 - Tous les effets indésirables apparus sous traitement rapportés chez ≥ 5 % des patients du bras lénalidomide/rituximab et à une fréquence (%) plus élevée d’au moins 2 % dans le bras lénalidomide par rapport au bras contrôle (population de sécurité).
o NHL-007 - Tous les effets indésirables apparus sous traitement de Grade 3 ou 4 rapportés chez ≥ 1 % des patients du bras lénalidomide/rituximab et avec une fréquence plus élevée d’au moins 1 % dans le bras lénalidomide par rapport au bras contrôle (population de sécurité).
o NHL-007 - Tous les effets indésirables graves apparus sous traitement rapportés chez ≥ 1 % des patients du bras lénalidomide/rituximab et avec une fréquence plus élevée d’au moins 1 % dans le bras lénalidomide par rapport au bras contrôle (population de sécurité).
Étude de phase 3 sur le lymphome folliculaire en un seul bras :
o NHL-008 - Tous les effets indésirables apparus sous traitement rapportés chez ≥ 5 % des patients.
o NHL-008 - Tous les effets indésirables apparus sous traitement de Grade 3 ou 4 rapportés chez ≥ 1 % des patients.
o NHL-008 - Tous les effets indésirables graves apparus sous traitement rapportés chez ≥ 1 % des patients.
◊ Événements indésirables rapportés comme graves dans les études cliniques menées dans le lymphome folliculaire.
+ S’applique uniquement aux effets indésirables graves.
* Le terme « éruption cutanée » inclut les TP rash et rash maculopapuleux.
** Le terme « leucopénie » inclut les TP leucopénie et globules blancs diminués.
*** Le terme « lymphopénie » inclut les TP lymphopénie et numération de lymphocytes diminuée.
Résumé tabulé des effets indésirables rapportés après la mise sur le marché
En plus des effets indésirables ci-dessus identifiés dans les études cliniques pivots, le tableau ci-dessous résulte des données collectées après la mise sur le marché.
Tableau 6. Effets indésirables rapportés après la mise sur le marché chez les patients traités par le lénalidomide
|
Classe de systèmes d’organes / Terme préférentiel |
Effets indésirables de tous Grades / Fréquence |
Effets indésirables de Grade 3 – 4 / Fréquence |
|
Infections et infestations |
Fréquence indéterminée Infections virales, y compris réactivation du virus de la varicelle et du zona et du virus de l’hépatite B |
Fréquence indéterminée Infections virales, y compris réactivation du virus de la varicelle et du zona et du virus de l’hépatite B |
|
Tumeurs malignes, bénignes et non précisées (incluant kystes et polypes) |
Rare Syndrome de lyse tumorale |
|
|
Affections du système immunitaire |
Rare Réaction anaphylactique^ Fréquence indéterminée Rejet d’une transplantation d’organe solide |
Rare Réaction anaphylactique^ |
|
Affections hématologiques et du système lymphatique |
Fréquence indéterminée Hémophilie acquise |
|
|
Affections endocriniennes |
Fréquent Hyperthyroïdie |
|
|
Affections respiratoires, thoraciques et médiastinales |
Peu fréquent Hypertension pulmonaire |
Rare Hypertension pulmonaire Fréquence indéterminée Pneumopathie interstitielle |
|
Affections gastro-intestinales |
Fréquence indéterminée Pancréatite, perforation gastro-intestinale (incluant perforation diverticulaire, intestinale et colique)^ |
|
|
Affections hépatobiliaires |
Fréquence indéterminée Insuffisance hépatique aiguë^, hépatite toxique^, hépatite cytolytique^, hépatite cholestatique^, hépatite mixte cytolytique/cholestatique^ |
Fréquence indéterminée Insuffisance hépatique aiguë^, hépatite toxique^ |
|
Affections de la peau et du tissu sous-cutané |
Peu fréquent Œdème de Quincke Rare Syndrome de Stevens Johnson^, Nécrolyse épidermique toxique^ Fréquence indéterminée Vascularite leucocytoclasique, réaction médicamenteuse accompagnée d’une éosinophilie et de symptômes systémiques^ (DRESS) |
^ Voir rubrique 4.8 Description de certains effets indésirables.
Description de certains effets indésirables
Tératogénicité
Le lénalidomide est structurellement proche du thalidomide. Le thalidomide est une substance active tératogène connue chez l’homme, provoquant des anomalies congénitales graves, potentiellement létales chez l’enfant à naître. Le lénalidomide induit chez les singes des malformations similaires à celles décrites avec le thalidomide (voir rubriques 4.6 et 5.3). Si le lénalidomide est pris pendant la grossesse, un effet tératogène du lénalidomide est attendu chez l’être humain.
Neutropénie et thrombopénie
· Myélome multiple non préalablement traité : patients recevant le lénalidomide en traitement d’entretien après une AGCS
Le lénalidomide en traitement d’entretien après une AGCS est associé à une fréquence plus élevée de neutropénie de Grade 4 par rapport au placebo en traitement d’entretien (32,1 % contre 26,7 % [16,1 % contre 1,8 % après le début du traitement d’entretien] dans l'étude CALGB 100104 et 16,4 % contre 0,7 % dans l'étude IFM 2005-02, respectivement). Des effets indésirables de type neutropénie apparus sous traitement entraînant l’arrêt du lénalidomide ont été rapportés chez 2,2 % des patients dans l'étude CALGB 100104 et 2,4 % des patients dans l'étude IFM 2005-02, respectivement. Des neutropénies fébriles de Grade 4 ont été rapportées, avec des fréquences similaires dans les bras lénalidomide en traitement d’entretien et placebo en traitement d’entretien dans les deux études (0,4 % contre 0,5 % [0,4 % contre 0,5 % après le début du traitement d’entretien] dans l'étude CALGB 100104 et 0,3 % contre 0 % dans l'étude IFM 2005-02, respectivement).
Le lénalidomide en traitement d’entretien après une AGCS est associé à une fréquence plus élevée de thrombopénie de Grade 3 ou 4 par rapport au placebo en traitement d’entretien (37,5 % contre 30,3 % [17,9 % contre 4,1 % après le début du traitement d’entretien] dans l'étude CALGB 100104 et 13,0 % contre 2,9 % dans l'étude IFM 2005-02, respectivement).
· Myélome multiple non préalablement traité : patients non éligibles à une greffe recevant le lénalidomide en association avec le bortézomib et la dexaméthasone
La fréquence des neutropénies de Grade 4 a été plus faible dans le bras lénalidomide en association avec le bortézomib et la dexaméthasone (RVd) que dans le bras comparateur Rd (2,7 % versus 5,9 %) de l’étude SWOG SO777.
La fréquence des neutropénies fébriles de Grade 4 a été similaire dans le bras RVd et dans le bras Rd (0,0 % versus 0,4 %).
La fréquence des thrombopénies de Grade 3 ou 4 a été plus élevée dans le bras RVd que dans le bras comparateur Rd (17,2 % versus 9,4 %).
· Myélome multiple non préalablement traité : patients non éligibles à une greffe, traités par le lénalidomide en association avec la dexaméthasone
L’association du lénalidomide avec la dexaméthasone chez les patients atteints de myélome multiple non préalablement traité est associée à une fréquence plus faible de neutropénie de Grade 4 (8,5 % pour Rd et Rd18) que MPT (15 %). Une neutropénie fébrile de Grade 4 a été observée peu fréquemment (0,6 % pour Rd et Rd18 contre 0,7 % pour MPT).
L’association du lénalidomide avec la dexaméthasone chez les patients atteints de myélome multiple non préalablement traité est associée à une fréquence plus faible de thrombopénie de Grade 3 et 4 (8,1 % pour Rd et Rd18) que MPT (11,1 %).
· Myélome multiple non préalablement traité : patients non éligibles à une greffe, traités par le lénalidomide en association avec le melphalan et la prednisone
L’association du lénalidomide avec le melphalan et la prednisone chez les patients atteints de myélome multiple non préalablement traité est associée à une fréquence plus élevée de neutropénie de Grade 4 (34,1 % pour MPR+R et MPR+p) que MPp+p (7,8 %). Une fréquence plus élevée de neutropénie fébrile de Grade 4 a été observée (1,7 % dans les bras MPR+R et MPR+p contre 0,0 % dans le bras MPp+p)
L’association du lénalidomide avec le melphalan et la prednisone chez les patients atteints de myélome multiple non préalablement traité est associée à une fréquence plus élevée de thrombopénie de Grade 3 et 4 (40,4 % pour MPR+R/MPR+p) que MPp+p (13,7 %).
· Myélome multiple : patients ayant reçu au moins un traitement antérieur
L’association du lénalidomide et de la dexaméthasone chez les patients atteints de myélome multiple est associée à une fréquence accrue de neutropénies de Grade 4 (5,1 % des patients traités par lénalidomide/dexaméthasone contre 0,6 % des patients traités par placebo/dexaméthasone). Des épisodes neutropéniques fébriles de Grade 4 ont plus rarement été observés (0,6 % des patients traités par lénalidomide/dexaméthasone contre 0,0 % des patients traités par placebo/ dexaméthasone).
L’association du lénalidomide et de la dexaméthasone chez les patients atteints de myélome multiple est associée à une incidence accrue des thrombopénies de Grade 3 et de Grade 4 (9,9 % et 1,4 %, respectivement, chez les patients traités par lénalidomide/dexaméthasone contre 2,3 % et 0,0 % chez les patients traités par placebo/dexaméthasone).
· Patients atteints de syndromes myélodysplasiques
Chez les patients atteints d’un syndrome myélodysplasique, le lénalidomide est associé à une fréquence accrue de neutropénie de Grade 3 ou 4 (74,6 % des patients traités par le lénalidomide versus 14,9 % des patients recevant le placebo dans l’étude de phase 3). Des épisodes de neutropénie fébrile de Grade 3 ou 4 ont été observés chez 2,2 % des patients traités par le lénalidomide versus 0,0 % des patients sous placebo.
Le lénalidomide est associé à une fréquence accrue de thrombopénie de Grade 3 ou 4 (37 % chez les patients traités par le lénalidomide versus 1,5 % des patients recevant le placebo dans l’étude de phase 3).
· Patients atteints de lymphome à cellules du manteau
Chez les patients atteints d’un lymphome à cellules du manteau, le lénalidomide est associé à une fréquence accrue de neutropénie de Grade 3 ou 4 (43,7 % des patients traités par le lénalidomide versus 33,7 % des patients du bras contrôle dans l’étude de phase 2). Des épisodes de neutropénie fébrile de Grade 3 ou 4 ont été observés chez 6,0 % des patients traités par le lénalidomide versus 2,4 % des patients du bras contrôle.
· Patients atteints de lymphome folliculaire
L’association du lénalidomide avec le rituximab chez les patients atteints d’un lymphome folliculaire, est associée à une fréquence plus élevée de neutropénies de Grade 3 ou 4 (50,7 % chez les patients traités par le lénalidomide/rituximab versus 12,2 % chez les patients recevant le placebo/rituximab). Toutes les neutropénies de Grade 3 ou 4 ont été réversibles après une interruption du traitement, une réduction de la dose et/ou l’administration de facteurs de croissance en traitement de support. En outre, une neutropénie fébrile a été observée peu fréquemment (2,7 % chez les patients traités par le lénalidomide/rituximab versus 0,7 % chez les patients recevant le placebo/rituximab).
L’association du lénalidomide avec le rituximab est également associée à une fréquence plus élevée des thrombopénies de Grade 3 ou 4 (1,4 % chez les patients traités par le lénalidomide/rituximab versus 0 % chez les patients recevant le placebo/rituximab).
Thrombo-embolie veineuse
Le risque de thrombose veineuse profonde (TVP) et d’embolie pulmonaire (EP) est majoré chez les patients atteints d’un myélome multiple traités par le lénalidomide en association avec la dexaméthasone, et, dans une moindre mesure, chez les patients traités par le lénalidomide en association avec le melphalan et la prednisone ou chez les patients présentant un myélome multiple, des syndromes myélodysplasiques ou un lymphome à cellules du manteau traités par le lénalidomide en monothérapie (voir rubrique 4.5).
L’administration concomitante d’érythropoïétine ou des antécédents de TVP peuvent également augmenter les risques de thrombose veineuse chez ces patients.
Infarctus du myocarde
Des cas d’infarctus du myocarde ont été rapportés chez les patients recevant du lénalidomide, particulièrement chez ceux qui présentent des facteurs de risque connus.
Troubles hémorragiques
Les troubles hémorragiques sont listés dans plusieurs catégories de classes d’organe : affections du sang et du système lymphatique ; affections du système nerveux (hémorragie intracrânienne) affections respiratoires, thoraciques et médiastinales (épistaxis) ; affections gastro-intestinales (saignement gingival, hémorragie hémorroïdaire, rectorragie) ; affections du rein et des voies urinaires (hématurie) ; lésions, intoxications et complications liées aux procédures (contusion) ; et affections vasculaires (ecchymoses).
Réactions allergiques et réactions cutanées sévères
Des cas de réactions allergiques, y compris d’angio-œdème et de réaction anaphylactique, et de réactions cutanées sévères, comme le syndrome de Steven-Johnson (SJS), la nécrolyse épidermique toxique (NET) et la réaction médicamenteuse accompagnée d’une éosinophilie et de symptômes systémiques (syndrome DRESS) ont été rapportées lors du traitement par le lénalidomide. Une réaction croisée possible entre le lénalidomide et le thalidomide a été rapportée dans la littérature. Les patients ayant des antécédents d’éruption sévère associée au traitement par le thalidomide ne doivent pas recevoir le lénalidomide (voir rubrique 4.4).
Cancers secondaires au traitement
Dans les essais cliniques menés chez des patients atteints d’un myélome, traités antérieurement avec l’association lénalidomide/dexaméthasone, comparés aux témoins, les cas consistant principalement en épithélioma basocellulaire ou spinocellulaire.
Leucémie aiguë myéloblastique
· Myélome multiple
Des cas de LAM ont été observés dans les études cliniques menées dans le myélome multiple non préalablement traité chez les patients traités par le lénalidomide en association avec le melphalan ou immédiatement après le MFD/AGCS (voir rubrique 4.4). Cette augmentation n’a pas été observée dans les études cliniques menées dans le myélome multiple non préalablement traité chez les patients recevant le lénalidomide en association avec la dexaméthasone par rapport aux patients recevant le thalidomide en association avec le melphalan et la prednisone.
· Syndromes myélodysplasiques
La présence au bilan initial d’anomalies cytogénétiques complexes ou d’une mutation de TP53 est associée à une progression en LAM chez les patients dépendants des transfusions et porteurs d’une anomalie Del (5q) (voir rubrique 4.4). Le risque estimé cumulé à 2 ans de progression en LAM était de 13,8 % chez les patients porteurs d’une anomalie Del (5q) isolée versus 17,3 % chez les patients porteurs de la Del (5q) et d’une autre anomalie cytogénétique et 38,6 % chez les patients présentant un caryotype complexe.
Dans une analyse post hoc d’une étude clinique du lénalidomide dans les syndromes myélodysplasiques, le taux estimé à 2 ans de progression en LAM était de 27,5 % chez les patients IHC-p53 positif et de 3,6 % chez les patients IHC-p53 négatif (p = 0,0038). Chez les patients IHC-p53 positif, un taux plus faible de progression en LAM a été observé chez ceux qui avaient obtenu une réponse d’indépendance transfusionnelle (IT) (11,1 %) que chez les non-répondeurs (34,8 %).
Affections hépatiques
Les effets indésirables suivants ont été rapportés après la mise sur le marché (fréquence indéterminée) : insuffisance hépatique aiguë et cholestase (potentiellement fatales toutes les deux), hépatite toxique, hépatite cytolytique et hépatite mixte cytolytique/cholestatique.
Rhabdomyolyse
De rares cas de rhabdomyolyse ont été observés, dont certains lorsque le lénalidomide est administré avec une statine.
Affections thyroïdiennes
Des cas d’hypothyroïdie et des cas d’hyperthyroïdie ont été rapportés (voir rubrique 4.4 Affections thyroïdiennes).
Réaction de poussée tumorale et syndrome de lyse tumorale
Dans l’étude MCL-002, environ 10 % des patients traités par le lénalidomide ont présenté une RPT versus 0 % dans le bras contrôle. Les événements sont survenus en majorité pendant le cycle 1, ils ont tous été évalués comme étant liés au traitement et étaient de Grade 1 ou 2 dans la majorité des cas. Les patients ayant un score MIPI élevé lors du diagnostic ou une charge tumorale élevée (au moins une lésion mesurant ≥ 7 cm dans le plus grand diamètre) avant le traitement peuvent présenter un risque de RPT. Dans l’étude MCL-002, un SLT a été rapporté chez un patient de chacun des deux bras de traitement. Dans l’étude supportive MCL-001, environ 10 % des patients ont présenté une RPT ; tous les cas étaient de sévérité de Grade 1 ou 2 et ils ont tous été évalués comme étant liés au traitement. Les événements sont survenus en majorité pendant le cycle 1. Il n’a pas été rapporté de cas de SLT dans l’étude MCL-001 (voir rubrique 4.4).
Dans l’étude NHL-007, une RPT a été rapportée chez 19 patients sur 146 (13,0 %) du bras lénalidomide/rituximab versus 1 patient sur 148 (0,7 %) dans le bras placebo/rituximab. La plupart des RPT (18 sur 19) rapportées dans le bras lénalidomide/rituximab se sont produites au cours des deux premiers cycles de traitement. Un patient du bras lénalidomide/rituximab atteint d’un lymphome folliculaire a présenté un événement de RPT de Grade 3 versus aucun patient dans le bras placebo/rituximab. Dans l’étude NHL-008, 7 patients sur 177 (4,0 %) patients atteints d’un lymphome folliculaire ont présenté une RPT ; (intensité de Grade 1 dans 3 cas et de Grade 2 dans 4 cas), 1 cas ayant été considéré comme grave. Dans l’étude NHL-007, un SLT est survenu chez 2 patients (1,4 %) atteints d’un lymphome folliculaire dans le bras lénalidomide/rituximab et chez aucun patient atteint d’un lymphome folliculaire dans le bras placebo/rituximab ; aucun patient n’a présenté d’événement de Grade 3 ou 4. Un SLT est survenu chez 1 patient (0,6 %) atteint d’un lymphome folliculaire dans l’étude NHL-008.
Cet événement unique a été jugé comme étant un effet indésirable grave, de Grade 3. En ce qui concerne l’étude NHL-007, aucun patient n’a dû interrompre le traitement par lénalidomide/rituximab en raison d’une RPT ou d’un SLT.
Affections gastro-intestinales
Des perforations gastro-intestinales ont été rapportées pendant le traitement par le lénalidomide. Les perforations gastro-intestinales peuvent entraîner des complications infectieuses et peuvent avoir une issue fatale.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/.
On ne dispose d’aucune expérience spécifique de la prise en charge d’un surdosage en lénalidomide, bien que certains patients des études d’évaluation de doses aient été exposés à des doses allant jusqu’à 150 mg et dans les études menées avec une seule dose, certains patients ont été exposés à des doses allant jusqu’à 400 mg. Le principal facteur de toxicité lors de ces études pouvant limiter la dose utilisée était d’ordre hématologique. En cas de surdosage, un traitement symptomatique sera conseillé.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : Immunosuppresseurs, autres immunosuppresseurs, Code ATC : L04AX04.
Mécanisme d’action
Le lénalidomide se lie directement à la protéine céréblon, une composante du complexe enzymatique ubiquitine ligase E3 de type cullin-RING constitué des protéines DDB1 (DNA [deoxyribonucleic acid] damage-binding protein 1), CUL4 (cullin 4) et Roc1 (regulator of cullins 1). Dans les cellules hématopoïétiques, la liaison du lénalidomide au céréblon entraîne le recrutement des protéines substrats Aiolos et Ikaros, des facteurs de transcription de la lignée lymphoïde, ce qui entraîne leur ubiquitination puis leur dégradation, avec pour résultat des effets cytotoxiques et immunomodulateurs directs.
En particulier, le lénalidomide inhibe la prolifération et favorise l’apoptose de certaines cellules malignes hématopoïétiques (y compris les plasmocytes malins de MM, les cellules malignes du lymphome folliculaire et les cellules présentant des délétions sur le chromosome 5), renforce l’immunité impliquant les lymphocytes T et les cellules tueuses naturelles (NK) et accroît le nombre des cellules NK, T et NKT. Dans les SMD del 5q, le lénalidomide inhibe sélectivement le clone anormal en augmentant l’apoptose des cellules présentant la délétion 5q.
L’association de lénalidomide et de rituximab augmente la cytotoxicité cellulaire dépendant des anticorps (ADCC) et favorise l’apoptose directe des cellules malignes du lymphome folliculaire.
Le mécanisme d’action du lénalidomide s’appuie également sur des propriétés anti-angiogènes et pro-érythropoïétiques. Le lénalidomide inhibe l’angiogenèse en bloquant la migration et l’adhésion des cellules endothéliales et la formation des micro-vaisseaux, amplifie la production d’hémoglobine fœtale par les cellules souches hématopoïétiques CD34+, et inhibe la production des cytokines pro-inflammatoires (TNF-α et IL-6, par exemple) par les monocytes.
Efficacité et sécurité clinique
L’efficacité et la sécurité de lénalidomide ont été évaluées dans six études de phase 3 menées dans le myélome multiple non préalablement traité, deux études de phase 3 menées dans le myélome multiple en rechute ou réfractaire, une étude de phase 3 et une étude de phase 2 menées dans les syndromes myélodysplasiques et une étude de phase 2 menée dans le lymphome à cellules du manteau, une étude de phase 3 et une étude de phase 3b menées dans les lymphomes non hodgkiniens indolents, qui sont décrites ci-dessous.
· Myélome multiple non préalablement traité
Lénalidomide en traitement d'entretien chez les patients ayant reçu une AGCS
L’efficacité et la sécurité du lénalidomide en traitement d’entretien ont été évaluées dans deux études de phase 3 multicentriques, randomisées en double aveugle, en deux bras, en groupes parallèles, contrôlées par placebo : CALGB 100104 et IFM 2005-02.
CALGB 100104
Des patients âgés de 18 à 70 ans présentant un myélome multiple symptomatique nécessitant un traitement et sans progression antérieure après un traitement initial étaient éligibles.
Les patients ont été randomisés selon un rapport 1:1 dans les 90 à 100 jours suivant l’AGCS, pour recevoir le lénalidomide ou le placebo en traitement d’entretien. La dose d’entretien était de 10 mg une fois par jour les jours 1 à 28 de chaque cycle de 28 jours (augmentée jusqu’à 15 mg une fois par jour après 3 mois en l’absence de toxicité dose-limitante) et le traitement était poursuivi jusqu’à la progression de la maladie.
Le critère d’évaluation principal de l'efficacité dans l'étude était la survie sans progression (SSP) (définie comme le délai entre la randomisation et la date de progression ou le décès, selon la première occurrence); l'étude ne disposait pas de la puissance nécessaire pour évaluer le critère de survie globale. Au total, 460 patients ont été randomisés : 231 patients sous lénalidomide et 229 sous placebo. Les caractéristiques démographiques et de la pathologie étaient comparables entre les deux bras.
L'aveugle de l'étude a été levé sur les recommandations du comité de surveillance des données, lorsque le seuil d'une analyse intermédiaire préprogrammée de la SSP a été dépassé. Après la levée de l'aveugle, les patients du bras placebo ont été autorisés à passer dans le bras lénalidomide avant la progression de leur maladie.
Les résultats de la SSP au moment de la levée de l'aveugle, suite à une analyse intermédiaire préprogrammée, avec gel des données le 17 décembre 2009 (suivi de 15,5 mois) ont montré une réduction de 62 % du risque de progression de la maladie ou de décès en faveur du lénalidomide (RR = 0,38 ; IC à 95 % 0,27, 0,54 ; p < 0,001). La SSP médiane globale était de 33,9 mois (IC à 95 % NE, NE) dans le bras lénalidomide contre 19,0 mois (IC à 95 % 16,2, 25,6) dans le bras placebo.
Le bénéfice pour la SSP a été observé aussi bien dans le sous-groupe de patients parvenus à une RC que dans le sous-groupe de patients non parvenus à une RC.
Les résultats de l’étude, selon une analyse actualisée au 1er février 2016 sont présentés dans le tableau 7.
Tableau 7. Synthèse des données d'efficacité globales
|
Lénalidomide (N = 231) |
Placebo (N = 229) |
|
|
SSP évaluée par les investigateurs |
||
|
SSP a médiane, mois (IC à 95 %)b |
56,9 (41,9 ; 71,7) |
29,4 (20,7 ; 35,5) |
|
RR [IC à 95 %]c ; valeur pd |
0,61 (0,48 ; 0,76) ; < 0,001 |
|
|
SSP2e |
||
|
SSP2 a médiane, mois (IC à 95 %)b |
80,2 (63,3 ; 101,8) |
52,8 (41,3 ; 64,0) |
|
RR [IC à 95 %]c ; valeur pd |
0,61 (0,48 ; 0,78) ; < 0,001 |
|
|
Survie globale |
||
|
SG a médiane, mois (IC à 95 %)b |
111,0 (101,8 ; NE) |
84,2 (71,0 ; 102,7) |
|
Taux de survie à 8 ans, % (ET) |
60,9 (3,78) |
44,6 (3,98) |
|
RR [IC à 95 %]c ; valeur pd |
0,61 (0,46 ; 0,81) ; < 0,001 |
|
|
Suivi |
||
|
Médianef (min., max.), mois : tous les patients survivants |
81,9 (0,0 ; 119,8) |
81,0 (4,1 ; 119,5) |
IC = intervalle de confiance ; RR = rapport de risque ; max. = maximum ; min. = minimum ; NE = non estimable ; SG = survie globale ; SSP = survie sans progression
a La médiane est basée sur l'estimation de Kaplan-Meier.
b IC à 95 % de la médiane.
c Basé sur un modèle pour risques proportionnels de Cox comparant les fonctions de risque associées aux bras de traitement indiqués.
d Valeur p déterminée par le test log-rank non stratifié des différences de la courbe de Kaplan-Meier entre les bras de traitement indiqués.
e Critère exploratoire (SSP2). Le lénalidomide reçu par les sujets du bras placebo qui ont changé de traitement avant la PM au moment de la levée de l'aveugle n’a pas été considéré comme un traitement de seconde intention.
f Suivi médian après une AGCS pour tous les sujets survivants.
Date de gel des données : 17 déc. 2009 et 1er fév. 2016.
IFM 2005-02
Des patients âgés de moins de 65 ans lors du diagnostic qui avaient reçu une AGCS et présentaient au moins une maladie stable au moment de la récupération hématologique étaient éligibles. Les patients ont été randomisés selon un rapport 1:1 pour recevoir le lénalidomide ou le placebo en traitement d'entretien (10 mg une fois par jour les jours 1 à 28 de cycles de 28 jours, augmentés jusqu'à 15 mg une fois par jour après 3 mois en l'absence de toxicité dose-limitante) après 2 cures de consolidation par le lénalidomide (25 mg/jour, les jours 1 à 21 d'un cycle de 28 jours). Le traitement devait être poursuivi jusqu'à la progression de la maladie.
Le critère principal d’évaluation était la SSP (définie comme le délai entre la randomisation et la date de progression ou le décès, selon la première occurrence); l'étude ne disposait pas de la puissance nécessaire pour évaluer le critère de survie globale. Au total, 614 patients ont été randomisés : 307 patients dans le bras lénalidomide et 307 dans le bras placebo.
L'aveugle de l'étude a été levé sur les recommandations du comité de surveillance des données, lorsque le seuil d'une analyse intermédiaire préprogrammée de la SSP a été dépassé. Après la levée de l’aveugle, les patients recevant le placebo ne sont pas passés dans le bras de traitement par lénalidomide avant la progression de leur maladie. Le bras lénalidomide a été interrompu par mesure de sécurité proactive, après avoir observé un déséquilibre dans l’apparition de cancers secondaires (voir rubrique 4.4).
Les résultats de la SSP au moment de la levée de l'aveugle, suite à une analyse intermédiaire préprogrammée, avec gel des données le 7 juillet 2010 (suivi de 31,4 mois) ont montré une réduction de 48 % du risque de progression de la maladie ou de décès en faveur du lénalidomide (RR = 0,52 ; IC à 95 % 0,41, 0,66 ; p < 0,001). La SSP médiane globale était de 40,1 mois (IC à 95 % 35,7, 42,4) dans le bras lénalidomide contre 22,8 mois (IC à 95 % 20,7, 27,4) dans le bras placebo.
Le bénéfice pour la SSP était moins important dans le sous-groupe de patients parvenus à une RC que dans le sous-groupe de patients non parvenus à une RC.
La SSP actualisée, avec gel des données le 1er février 2016 (suivi de 96,7 mois) continue à montrer un avantage en SSP : RR = 0,57 (IC à 95 % 0,47, 0,68 ; p < 0,001). La SSP médiane globale était de 44,4 mois (IC à 95 % 39,6, 52,0) dans le bras lénalidomide contre 23,8 mois (IC à 95 % 21,2, 27,3) dans le bras placebo. Pour la SSP2, le RR observé était de 0,80 (IC à 95 % 0,66, 0,98 ; p = 0,026) pour le lénalidomide par rapport au placebo. La SSP2 médiane globale était de 69,9 mois (IC à 95 % 58,1, 80,0) dans le bras lénalidomide contre 58,4 mois (IC à 95 % 51,1, 65,0) dans le bras placebo. Pour la SG, le RR observé était de 0,90 (IC à 95 % 0,72, 1,13 ; p = 0,355) pour le lénalidomide par rapport au placebo. La survie médiane globale était de 105,9 mois (IC à 95 % 88,8, NE) dans le bras lénalidomide contre 88,1 mois (IC à 95 % 80,7, 108,4) dans le bras placebo.
Lénalidomide en association avec le bortézomib et la dexaméthasone chez les patients non éligibles à une greffe de cellules souches
L’étude SWOG S0777 visait à évaluer l’ajout du bortézomib à l’association de base de lénalidomide et dexaméthasone en traitement initial, suivi du traitement par l’association Rd poursuivi jusqu’à la progression de la maladie, chez des patients atteints d’un myélome multiple non préalablement traité qui n’étaient pas éligibles à une greffe ou qui étaient éligibles à une greffe mais chez lesquels une greffe n’était pas prévue immédiatement.
Les patients du bras lénalidomide, bortézomib et dexaméthasone (RVd) ont reçu le lénalidomide 25 mg/jour par voie orale les jours 1 à 14, le bortézomib 1,3 mg/m2 par voie intraveineuse les jours 1, 4, 8 et 11 et la dexaméthasone 20 mg/jour par voie orale les jours 1, 2, 4, 5, 8, 9, 11 et 12 de chaque cycle de 21 jours répétés pour atteindre un total allant jusqu’à 8 cycles de 21 jours (24 semaines). Les patients du bras lénalidomide et dexaméthasone (Rd) ont reçu le lénalidomide 25 mg/jour par voie orale les jours 1 à 21 et la dexaméthasone 40 mg/jour par voie orale les jours 1, 8, 15 et 22 de chaque cycle de 28 jours répétés pour un total allant jusqu’à 6 cycles de 28 jours (24 semaines). Les patients des deux bras continuaient à recevoir l’association Rd : le lénalidomide 25 mg/jour par voie orale les jours 1 à 21 et la dexaméthasone 40 mg/jour par voie orale les jours 1, 8, 15 et 22 de chaque cycle de 28 jours. Le traitement devait être poursuivi jusqu’à la progression de la maladie.
Le critère d’efficacité principal dans l’étude était la survie sans progression (SSP). Au total, 523 patients ont été inclus dans l’étude, dont 263 patients randomisés dans le bras RVd et 260 patients randomisés dans le bras Rd. Les caractéristiques démographiques et pathologiques initiales des patients étaient bien équilibrées entre les bras.
Les résultats de SSP, selon l’évaluation de l’IRAC, au moment de l’analyse principale avec gel des données au 5 novembre 2015 (50,6 mois de suivi) ont montré une réduction de 24 % du risque de progression de la maladie ou de décès en faveur de l’association RVd (RR = 0,76 ; IC à 95 % 0,61 ; 0,94 ; p = 0,010). La SSP médiane globale était 42,5 mois (IC à 95 % 34,0, 54,8) dans le bras RVd contre 29,9 mois (IC à 95 % 25,6, 38,2) dans le bras Rd. Le bénéfice a été observé quelle que soit l’éligibilité à une greffe de cellules souches.
Les résultats de l’étude, avec gel des données le 1er décembre 2016, dans laquelle la durée médiane de suivi de l’ensemble des patients en vie était de 69,0 mois, sont présentés dans le tableau 8. Le bénéfice en faveur de l’association RVd a été observé quelle que soit l’éligibilité à une greffe de cellules souches.
Tableau 8. Synthèse des données d’efficacité globales
|
|
Traitement initial |
|
|
RVd (8 cycles de 3 semaines) (N = 263) |
Rd (6 cycles de 4 semaines) (N = 260) |
|
|
SSP évaluée par l’IRAC (mois) |
||
|
SPP médianea, mois (IC à 95 %)b |
41,7 (33,1, 51,5) |
29,7 (24,2, 37,8) |
|
RR [IC à 95 %]c ; valeur pd |
0,76 (0,62, 0,94); 0,010 |
|
|
Survie globale (mois) |
||
|
SG médianea, mois (IC à 95 %)b |
89,1 (76,1, NE) |
67,2 (58,4, 90,8) |
|
RR [IC à 95 %]c ; valeur pd |
0,72 (0,56, 0,94); 0,013 |
|
|
Réponse – n (%) |
||
|
Réponse globale: RC, TBRP, ou RP |
199 (75,7) |
170 (65,4) |
|
≥ TBRP |
153 (58,2) |
83 (31,9) |
|
Suivi (mois) |
||
|
Médianee (min, max): tous patients |
61,6 (0,2, 99,4) |
59,4 (0,4, 99,1) |
IC = intervalle de confiance ; RR = rapport de risque ; max = maximum ; min = minimum ; NE = non estimable ; SG = survie globale ; SSP = survie sans progression
a La médiane est basée sur l’estimation de Kaplan-Meier.
b IC à 95 % bilatéral de la médiane.
c Basé sur un modèle pour risques proportionnels de Cox comparant les fonctions de risque associées aux bras de traitement (RVd:Rd).
d Valeur p déterminée par le test log-rank non stratifié.
e Durée médiane de suivi calculée à partir de la date de randomisation.
Date de gel des données : 1er décembre 2016.
Les résultats de SG actualisés, avec gel des données le 1er mai 2018 (durée médiane de suivi de 84,2 mois pour les patients survivants), continuent à montrer un avantage de SG en faveur de l’association RVd : RR = 0,73 (IC à 95 % 0,57 ; 0,94 ; p = 0,014). Les pourcentages de patients en vie après 7 ans étaient de 54,7 % dans le bras RVd contre 44,7 % dans le bras Rd.
Lénalidomide administré en association avec la dexaméthasone chez les patients non éligibles à une greffe de cellules souches
La sécurité et l’efficacité du lénalidomide ont été évaluées dans une étude de phase 3 multicentrique, randomisée en ouvert, en trois bras (MM-020), menée chez des patients âgés de 65 ans ou plus ou chez les patients âgés de moins de 65 ans, qui n’étaient pas éligibles à une greffe de cellules souches parce qu’ils l’avaient refusée ou ne pouvaient pas en bénéficier en raison du coût ou pour d’autres motifs. L’étude (MM-020) visait à comparer l’association de lénalidomide et dexaméthasone (Rd) administrée pendant deux durées différentes (jusqu’à la progression de la maladie [bras Rd] ou pendant un maximum de 18 cycles de 28 jours [72 semaines, bras Rd18] à l’association de melphalan, prednisone et thalidomide (MPT) administrée pendant un maximum de 12 cycles de 42 jours (72 semaines).
Les patients ont été randomisés (1:1:1) dans l’un des trois bras de traitement. Les patients ont été stratifiés au moment de la randomisation en fonction de l'âge (≤ 75 ans versus > 75 ans), du stade (stades ISS I et II versus stade III) et du pays.
Les patients des bras Rd et Rd18 ont pris 25 mg de lénalidomide une fois par jour les jours 1 à 21 des cycles de 28 jours conformément au bras de l’étude. La dexaméthasone 40 mg était prise une fois par jour les jours 1, 8, 15 et 22 de chaque cycle de 28 jours. Dans les bras Rd et Rd18, la dose initiale et le schéma posologique étaient ajustés en fonction de l'âge et de la fonction rénale (voir rubrique 4.2). Les patients âgés de plus de 75 ans ont reçu une dose de 20 mg de dexaméthasone une fois par jour les jours 1, 8, 15 et 22 de chaque cycle de 28 jours. Tous les patients ont reçu une thromboprophylaxie (héparine de bas poids moléculaire, warfarine, héparine, aspirine à faible dose) pendant l’étude.
Le critère d’efficacité principal dans l’étude était la survie sans progression (SSP). Au total, 1 623 patients ont été inclus dans l’étude, dont 535 patients randomisés dans le bras Rd, 541 patients dans le bras Rd18 et 547 patients dans le bras MPT. Les caractéristiques démographiques et pathologiques initiales des patients étaient bien équilibrées dans les trois bras. En général, les patients de l’étude présentaient une maladie de stade avancé : sur la population totale de l’étude, 41 % présentaient une maladie de stade ISS III et 9 % une insuffisance rénale sévère (clairance de la créatinine [ClCr] < 30 mL/min). L'âge médian était de 73 ans dans les trois bras.
Les résultats de l’étude selon une analyse actualisée de la SSP, de la SSP2 et de la SG avec gel des données le 3 mars 2014 dans laquelle la durée médiane de suivi de l’ensemble des patients survivants était de 45,5 mois sont présentés dans le tableau 9.
Tableau 9. Synthèse des données d'efficacité globales
|
Rd |
Rd18 |
MPT |
||
|
(N = 535) |
(N = 541) |
(N = 547) |
||
|
Survie sans progression évaluée par les investigateurs – (mois) |
||||
|
SSP médianea, mois (IC à 95 %)b |
26,0 (20,7, 29,7) |
21,0 (19,7, 22,4) |
21,9 (19,8, 23,9) |
|
|
RR [IC à 95 %]c ; valeur pd |
||||
|
Rd vs MPT |
0,69 (0,59, 0,80) ; < 0,001 |
|||
|
Rd vs Rd18 |
0,71 (0,61, 0,83) ; < 0,001 |
|||
|
Rd18 vs MPT |
0,99 (0,86, 1,14) ; 0,866 |
|||
|
SSP2e − (mois) |
||||
|
SSP2 médianea, mois (IC à 95 %)b |
42,9 (38,1, 47,4) |
40,0 (36,2, 44,2) |
35,0 (30,4, 37,8) |
|
|
RR [IC à 95 %]c ; valeur pd |
||||
|
Rd vs MPT |
0,74 (0,63, 0,86) ; < 0,001 |
|||
|
Rd vs Rd18 |
0,92 (0,78, 1,08) ; 0,316 |
|||
|
Rd18 vs MPT |
0,80 (0,69, 0,93) ; 0,004 |
|||
|
Survie globale (mois) |
||||
|
SG médianea, mois (IC à 95 %)b |
58,9 (56,0, NE) |
56,7 (50,1, NE) |
48,5 (44,2, 52,0) |
|
|
RR [IC à 95 %]c ; valeur pd |
||||
|
Rd vs MPT |
0,75 (0,62, 0,90) ; 0,002 |
|||
|
Rd vs Rd18 |
0,91 (0,75, 1,09) ; 0,305 |
|||
|
Rd18 vs MPT |
0,83 (0,69, 0,99) ; 0,034 |
|||
|
Suivi (mois) |
||||
|
Médianef (min, max) : tous patients |
40,8 (0,0, 65,9) |
40,1 (0,4, 65,7) |
38,7 (0,0, 64,2) |
|
|
Taux de réponse globale g, n (%) |
||||
|
RC |
81 (15,1) |
77 (14,2) |
51 (9,3) |
|
|
TBRP |
152 (28,4) |
154 (28,5) |
103 (18,8) |
|
|
RP |
169 (31,6) |
166 (30,7) |
187 (34,2) |
|
|
Réponse globale : RC, TBRP ou RP |
402 (75,1) |
397 (73,4) |
341 (62,3) |
|
|
Durée de la réponse – (mois)h |
||||
|
Médianea (IC à 95 %)b |
35,0 (27,9, 43,4) |
22,1 (20,3, 24,0) |
22,3 (20,2, 24,9) |
|
TAM = traitement antimyélome ; IC = intervalle de confiance ; RC = réponse complète ; d = dexaméthasone à faible dose ; RR = rapport de risque ; IMWG = International Myeloma Working Group ; IRAC = Independent Response Adjudication Committee (comité indépendant d’évaluation de la réponse) ; M = melphalan ; max = maximum ; min = minimum ; NE = non estimable ; SG = survie globale ; P = prednisone ; SSP =survie sans progression ; RP = réponse partielle ; R = lénalidomide ; Rd = Rd administrés jusqu’à la documentation de la progression de la maladie ; Rd18 = Rd administrés pendant ≤ 18 cycles ; ES = erreur standard ; T = thalidomide ; TBRP = très bonne réponse partielle ; vs = versus.
a La médiane est basée sur l’estimation de Kaplan-Meier.
b IC à 95 % de la médiane.
c Basé sur un modèle pour risques proportionnels de Cox comparant les fonctions de risque associées aux bras de traitement indiqués.
d Valeur p déterminée par le test log-rank non stratifié des différences de la courbe de Kaplan-Meier entre les bras de traitement indiqués.
e Critère exploratoire (SSP2).
f La médiane est la statistique univariée sans ajustement pour censure.
g Meilleure réponse évaluée par le comité indépendant pendant la phase de traitement de l’étude (pour les définitions de chaque catégorie de réponse, date de gel des données : 24 mai 2013).
h Date de gel des données : 24 mai 2013.
Lénalidomide en association avec le melphalan et la prednisone suivis d’un traitement d’entretien chez les patients non éligibles à une greffe
La sécurité et l’efficacité du lénalidomide ont été évaluées dans une étude de phase 3 multicentrique randomisée en double aveugle, contrôlée par placebo en trois bras (MM-015), menée chez des patients qui étaient âgés de 65 ans et plus et qui avaient une créatininémie < 2,5 mg/dL. L’étude visait à comparer l’association de lénalidomide avec le melphalan et la prednisone (MPR) avec ou sans traitement d’entretien par le lénalidomide jusqu’à la progression de la maladie à l’association de melphalan et prednisone administrée pendant 9 cycles au maximum. Les patients ont été randomisés selon un rapport 1:1:1 dans l’un des trois bras de traitement. Les patients ont été stratifiés au moment de la randomisation en fonction de l'âge (≤ 75 ans versus > 75 ans) et du stade (ISS ; stades I et II versus stade III).
Cette étude visait à évaluer l’utilisation de l’association MPR (melphalan 0,18 mg/kg par voie orale les jours 1 à 4 de chaque cycle de 28 jours, prednisone 2 mg/kg par voie orale les jours 1 à 4 de chaque cycle de 28 jours et lénalidomide 10 mg/jour par voie orale les jours 1 à 21 de chaque cycle de 28 jours) en traitement d’induction pendant 9 cycles au maximum. Les patients qui avaient terminé les 9 cycles ou qui ne pouvaient pas les terminer en raison d’une toxicité ont poursuivi par un traitement d’entretien débutant avec 10 mg de lénalidomide par voie orale les jours 1 à 21 de chaque cycle de 28 jours jusqu’à la progression de la maladie.
Le critère d’efficacité principal dans l’étude était la survie sans progression (SSP). Au total, 459 patients ont été inclus dans l’étude, dont 152 patients randomisés dans le bras MPR+R, 153 patients dans le bras MPR+p et 154 patients dans le bras MPp+p. Les caractéristiques démographiques et pathologiques initiales des patients étaient bien équilibrées entre les trois bras ; en particulier, environ 50 % des patients inclus dans chaque bras présentaient les caractéristiques suivantes : stade ISS III et clairance de la créatinine < 60 mL/min. L'âge médian était de 71 ans dans les bras MPR+R et MPR+p et de 72 ans dans le bras MPp+p.
Les résultats de l’étude selon une analyse de la SSP, de la SSP2 et de la SG avec gel des données en avril 2013 dans laquelle la durée médiane de suivi de l’ensemble des patients survivants était de 62,4 mois sont présentés dans le tableau 10.
Tableau 10. Synthèse des données d’efficacité globales
|
MPR+R (N = 152) |
MPR+p (N = 153) |
MPp +p (N = 154) |
|||
|
Survie sans progression évaluée par les investigateurs – (mois) |
|||||
|
SSP médiane a, mois (IC à 95 %) |
27,4 (21,3, 35,0) |
14,3 (13,2, 15,7) |
13,1 (12,0, 14,8) |
||
|
RR [IC à 95 %], valeur p |
|||||
|
MPR+R vs MPp+p |
0,37 (0,27, 0,50) ; < 0,001 |
||||
|
MPR+R vs MPR+p |
0,47 (0,35, 0,65) ; < 0,001 |
||||
|
MPR+p vs MPp +p |
0,78 (0,60, 1,01) ; 0,059 |
||||
|
SSP2 − (mois)¤ |
|||||
|
SSP2 médiane a, mois (IC à 95 %) |
39,7 (29,2, 48,4) |
27,8 (23,1, 33,1) |
28,8 (24,3, 33,8) |
||
|
RR [IC à 95 %] ; valeur p |
|||||
|
MPR+R vs MPp+p |
0,70 (0,54, 0,92) ; 0,009 |
||||
|
MPR+R vs MPR+p |
0,77 (0,59, 1,02) ; 0,065 |
||||
|
MPR+p vs MPp +p |
0,92 (0,71, 1,19) ; 0,051 |
||||
|
Survie globale (mois) |
|||||
|
SG médiane a, mois (IC à 95 %) |
55,9 (49,1, 67,5) |
51,9 (43,1, 60,6) |
53,9 (47,3, 64,2) |
||
|
RR [IC à 95 %] ; valeur p |
|||||
|
MPR+R vs MPp+p |
0,95 (0,70, 1,29) ; 0,736 |
||||
|
MPR+R vs MPR+p |
0,88 (0,65, 1,20) ; 0,43 |
||||
|
MPR+p vs MPp +p |
1,07 (0,79, 1,45) ; 0,67 |
||||
|
Suivi (mois) |
|||||
|
Médiane (min, max) : tous les patients |
48,4 (0,8, 73,8) |
46,3 (0,5, 71,9) |
50,4 (0,5, 73,3) |
||
|
Taux de réponse globale évaluée par les investigateurs, n (%) |
|
|
|
|
|
|
RC |
30 (19,7) |
17 (11,1) |
9 (5,8) |
||
|
RP |
90 (59,2) |
99 (64,7) |
75 (48,7) |
||
|
Maladie stable (MS) |
24 (15,8) |
31 (20,3) |
63 (40,9) |
||
|
Réponse non évaluable (NE) |
8 (5,3) |
4 (2,6) |
7 (4,5) |
||
|
Durée de la réponse évaluée par les investigateurs (RC + RP) − (mois) |
|||||
|
Médianea (IC à 95 %) |
26,5 (19,4, 35,8) |
12,4 (11,2, 13,9) |
12,0 (9,4, 14,5) |
||
IC = intervalle de confiance ; RC = réponse complète ; RR = rapport de risque ; M = melphalan ; NE = non estimable ; SG = survie globale ; p = placebo ; P = prednisone ; MP = maladie en progression ; RP = réponse partielle ; R = lénalidomide ; MS = maladie stable ; TBRP = très bonne réponse partielle
a La médiane est basée sur l’estimation de Kaplan-Meier.
¤ La SSP2 (un critère exploratoire) était définie pour tous les patients randomisés (population ITT) comme le délai entre la randomisation et le début d’un traitement antimyélome (TAM) de 3e ligne ou le décès.
Études de confirmation dans le myélome multiple non préalablement traité
Une étude de phase 3 multicentrique, randomisée, en ouvert, (ECOG E4A03) a été menée chez 445 patients atteints d’un myélome multiple non préalablement traité ; 222 patients ont été randomisés dans le bras lénalidomide/dexaméthasone à faible dose et 223 patients dans le bras lénalidomide/dexaméthasone à dose standard. Les patients randomisés dans le bras lénalidomide/dexaméthasone à dose standard ont reçu 25 mg par jour de lénalidomide des jours 1 à 21 de cycles de 28 jours et 40 mg par jour de dexaméthasone les jours 1 à 4, 9 à 12 et 17 à 20 pendant les quatre premiers cycles de 28 jours. Les patients randomisés dans le bras lénalidomide/dexaméthasone à faible dose ont reçu 25 mg par jour de lénalidomide des jours 1 à 21 de cycles de 28 jours et une dose faible de dexaméthasone (40 mg par jour les jours 1, 8, 15 et 22 de chaque cycle de 28 jours). Dans le bras lénalidomide/dexaméthasone à faible dose, 20 patients (9,1 %) ont eu au moins une interruption de traitement contre 65 patients (29,3 %) dans le bras lénalidomide/dexaméthasone à dose standard.
Une analyse post hoc a montré une mortalité plus faible dans le bras lénalidomide/ dexaméthasone à faible dose (6,8 %, 15/220) que dans le bras lénalidomide/dexaméthasone à dose standard (19,3 %, 43/223) dans la population de patients atteints d’un myélome multiple non préalablement traité, avec un suivi médian de 72,3 semaines.
Cependant, avec un suivi plus long, la différence de survie globale en faveur de l’association lénalidomide/dexaméthasone à faible dose a tendance à diminuer.
· Myélome multiple chez les patients ayant reçu au moins un traitement antérieur
L’efficacité et la sécurité d’emploi du lénalidomide ont été évaluées lors de deux études de phase 3, multicentriques, randomisées, en double aveugle, contrôlées par placebo, en groupes parallèles (MM-009 et MM-010), ayant comparé l’association lénalidomide/dexaméthasone à la dexaméthasone en monothérapie chez des patients ayant déjà été traités, atteints de myélome multiple. Parmi les 353 patients des études MM-009 et MM-010 ayant reçu l’association lénalidomide/dexaméthasone, 45,6 % étaient âgés de 65 ans ou plus. Sur l’ensemble des 704 patients évalués lors des études MM-009 et MM-010, 44,6 % étaient âgés de 65 ans ou plus.
Lors des deux études, les patients du groupe lénalidomide/dexaméthasone (lén/dex) ont pris 25 mg de lénalidomide par voie orale en une prise par jour les jours 1 à 21 et une gélule de placebo correspondante en une prise par jour les jours 22 à 28 de chaque cycle de 28 jours. Les patients du groupe placebo/dexaméthasone (placebo/dex) ont pris 1 gélule placebo les jours 1 à 28 de chaque cycle de 28 jours. Les patients des deux groupes ont pris 40 mg de dexaméthasone par voie orale en une prise par jour les jours 1 à 4, 9 à 12 et 17 à 20 de chaque cycle de 28 jours pendant les 4 premiers cycles de 28 jours. La dose de dexaméthasone a ensuite été réduite à 40 mg par voie orale en une prise par jour les jours 1 à 4 des cycles suivants de 28 jours après les 4 premiers cycles du traitement. Lors des deux études, le traitement devait être poursuivi jusqu’à la progression de la maladie. Dans les deux études, des ajustements posologiques étaient autorisés en fonction des résultats cliniques et des analyses biologiques.
Le critère d’évaluation principal d’efficacité des deux études était le temps jusqu’à progression (time to progression, TTP). Au total, 353 patients ont été évalués dans le cadre de l’étude MM-009, 177 dans le groupe lén/dex et 176 dans le groupe placebo/dex et, au total, 351 patients ont été évalués dans le cadre de l’étude MM-010, 176 dans le groupe lén/dex et 175 dans le groupe placebo/dex.
Lors des deux études, les caractéristiques démographiques et pathologiques initiales des patients étaient comparables dans les deux groupes (lén/dex et placebo/dex). Les populations étudiées présentaient dans les deux cas un âge médian de 63 ans, avec une proportion hommes/femmes comparable. Les indices de performance tels que définis par l’ECOG (Eastern Cooperative Oncology Group) étaient comparables dans les deux groupes, comme l’étaient le nombre et le type des traitements antérieurs.
Les analyses intermédiaires préalablement planifiées pour les deux études ont montré que l’association lén/dex donnait un résultat significativement supérieur en termes statistiques (p < 0,00001) à celui obtenu par la dexaméthasone en monothérapie sur le critère principal d’évaluation de l’efficacité, le TTP (suivi médian de 98,0 semaines). Les taux de réponses complètes et de réponses globales lén/dex ont également été significativement supérieurs à ceux du groupe placebo/dex dans les deux études. Les résultats de ces analyses ont eu pour conséquence la levée de l’aveugle, dans les deux études, de façon à ce que les patients du groupe placebo/dex puissent bénéficier du traitement par l’association lén/dex.
Une analyse de l’efficacité après un suivi prolongé a été menée avec un suivi médian de 130,7 semaines. Le tableau 11 récapitule les résultats d’efficacité issus des analyses de suivi des études MM-009 et MM-010 groupées.
Dans cette analyse du suivi prolongé, le TTP médian était de 60,1 semaines (IC à 95 % : 44,3 – 73,1) chez les patients traités par lén/dex (N = 353) contre 20,1 semaines (IC à 95 % : 17,7 – 20,3) chez les patients traités par placebo/dex (N = 351). La durée médiane de survie sans progression de la maladie a été de 48,1 semaines (IC à 95 % : 36,4 – 62,1) chez les patients traités par lén/dex contre 20,0 semaines (IC à 95 % : 16,1 – 20,1) chez les patients traités par placebo/dex. La durée médiane de traitement a été de 44,0 semaines (min : 0,1 ; max : 254,9) dans le bras lén/dex, et de 23,1 semaines (min : 0,3 ; max : 238,1) dans le bras placebo/dex. Dans les deux études, les taux de réponses complètes (RC), de réponses partielles (RP) et de réponses globales (RC+RP) sont restés significativement plus élevés dans le groupe lén/dex que dans le groupe placebo/dex.
Le taux médian de survie globale dans l’analyse du suivi prolongé des études groupées est de 164,3 semaines (IC à 95 % : 145,1 – 192,6) chez les patients traités par lén/dex contre 136,4 semaines (IC à 95 % : 113,1 – 161,7) chez les patients ayant reçu l’association placebo/dex.
Bien que 170 des 351 patients randomisés dans le groupe placebo/dex aient finalement reçu le traitement par le lénalidomide après progression de la maladie ou une fois l’insu levé, l’analyse groupée de la survie globale a permis de montrer une différence statistiquement significative concernant la survie en faveur de l’association lén/dex par rapport à l’association placebo/dex (RR = 0,833 ; IC à 95 % : 0,687 – 1,009 ; p = 0,045).
Tableau 11. Résumé des résultats des analyses de l’efficacité à la date de fin de collecte des données après suivi prolongé — études MM-009 et MM-010 regroupées (dates de fin de collecte des données respectives, 23 juillet 2008 et 2 mars 2008)
|
Critère d’évaluation |
lén/dex |
placebo/dex |
|
|
|
(N = 353) |
(N = 351) |
|
|
Délai avant événement |
|
|
RR [IC à 95 %], pa |
|
Temps jusqu’à progression |
60,1
|
20,1
|
0,350 [0,287, 0,426], |
|
médiane [IC à 95 %], semaines |
[44,3 ; 73,1] |
[17,7 ; 20,3] |
p < 0,001 |
|
Survie sans progression |
48,1
|
20,0
|
0,393 [0,326 ; 0,473],
|
|
médiane [IC à 95 %], semaines |
[36,4 ; 62,1] |
[16,1 ; 20,1] |
p < 0,001 |
|
Survie globale |
164,3 |
136,4 |
0,833 [0,687 ; 1,009], |
|
médiane [IC à 95 %], semaines |
[145,1 ; 192,6]
|
[113,1 ; 161,7]
|
p = 0,045 |
|
Taux de survie globale à 1 an |
82 % |
75 % |
|
|
Taux de réponse |
Odds ratio [IC à 95 %], pb |
||
|
Réponses globales [n, %] |
212 (60,1) |
75 (21,4) |
5,53 [3,97 ; 7,71], p < 0,001 |
|
Réponses complètes [n, %] |
58 (16,4) |
11 (3,1) |
6,08 [3,13 ; 11,80], p < 0,001 |
a : Test bilatéral du log-rank comparant les courbes de survie entre les groupes de traitement.
b : Test bilatéral du χ² corrigé en fonction de la continuité.
· Syndromes myélodysplasiques
L’efficacité et la tolérance du lénalidomide ont été évaluées dans deux études principales menées chez des patients présentant une anémie avec dépendance transfusionnelle due à un syndrome myélodysplasique à risque faible ou intermédiaire 1 associé à une anomalie cytogénétique de type délétion 5q, avec ou sans autres anomalies cytogénétiques : une étude de phase 3 multicentrique, randomisée en double aveugle, placebo contrôle, de trois bras, évaluant deux doses orales de lénalidomide (10 mg et 5 mg) versus placebo (MDS-004) et une étude de phase 2 multicentrique du traitement en ouvert par le lénalidomide (10 mg) uniquement (MDS-003).
Les résultats présentés ci-dessous représentent la population en intention-de-traiter des études MDS-003 et MDS-004 ; les résultats dans la sous-population de patients porteurs de l’anomalie Del 5q isolée sont également présentés séparément.
Dans l’étude MDS-004, 205 patients ont été randomisés, selon un rapport égal, pour recevoir le lénalidomide 10 mg, le lénalidomide 5 mg ou le placebo : le critère principal d’efficacité consistait en une comparaison des taux de réponse d’indépendance transfusionnelle dans les bras lénalidomide 10 mg et 5 mg par rapport au bras placebo (phase en double aveugle de 16 à 52 semaines et extension en ouvert allant jusqu’à 156 semaines au total). Le traitement devait être arrêté chez les patients qui ne présentaient pas au moins une réponse érythroïde mineure après 16 semaines.
Les patients présentant une réponse érythroïde au moins mineure pouvaient poursuivre le traitement jusqu’à la rechute, la progression de la maladie ou une toxicité inacceptable. Les patients qui avaient reçu initialement le placebo ou le lénalidomide 5 mg et qui n’avaient pas obtenu une réponse érythroïde au moins mineure après 16 semaines de traitement pouvaient passer du placebo au lénalidomide 5 mg ou poursuivre le traitement par le lénalidomide à une dose plus élevée (de 5 mg à 10 mg).
Dans l’étude MDS-003, au cours de laquelle 148 patients ont reçu le lénalidomide à la dose de 10 mg, l’analyse du critère principal consistait à évaluer l’efficacité du traitement par le lénalidomide sur l’amélioration hématopoïétique chez les patients présentant un syndrome myélodysplasique à risque faible ou intermédiaire 1.
Tableau 12. Synthèse des résultats d’efficacité – Études MDS-004 (phase en double aveugle) et MDS-003, population en intention-de-traiter
|
Critère d’évaluation |
MDS-004 N = 205 |
MDS-003 N = 148 |
||
|
10 mg† N = 69 |
5 mg†† N = 69 |
Placebo* N = 67 |
10 mg N = 148 |
|
|
Indépendance transfusionnelle (≥ 182 jours) # |
38 (55,1 %) |
24 (34,8 %) |
4 (6,0 %) |
86 (58,1 %) |
|
Indépendance transfusionnelle (≥ 56 jours) # |
42 (60,9 %) |
33 (47,8 %) |
5 (7,5 %) |
97 (65,5 %) |
|
Délai médian jusqu’à l’indépendance transfusionnelle (semaines) |
4,6 |
4,1 |
0,3 |
4,1 |
|
Durée médiane d’indépendance transfusionnelle (semaines) |
NR∞ |
NR |
NR |
114,4 |
|
Augmentation médiane de l’Hb, g/dL |
6,4 |
5,3 |
2,6 |
5,6 |
† Patients traités par le lénalidomide 10 mg pendant 21 jours de cycles de 28 jours.
†† Patients traités par le lénalidomide 5 mg pendant 28 jours de cycles de 28 jours.
La majorité des patients sous placebo a arrêté le traitement en double aveugle après 16 semaines en raison d’un manque d’efficacité avant d’entrer dans la phase en ouvert.
# Associée à une augmentation de l’Hb ≥ 1g/dL
∞ Non atteinte (la médiane n’a pas été atteinte).
Dans l’étude MDS-004, le pourcentage de patients atteints d’un syndrome myélodysplasique ayant atteint le critère principal d’indépendance transfusionnelle (> 182 jours) a été significativement plus élevé avec le lénalidomide 10 mg qu’avec le placebo (55,1 % versus 6,0 %). Chez les 47 patients porteurs d’une anomalie cytogénétique del 5q isolée et traités par le lénalidomide 10 mg, 27 patients (57,4 %) ont obtenu l’indépendance vis-à-vis des transfusions de globules rouges.
Dans le bras lénalidomide 10 mg, le délai médian jusqu’à l’indépendance transfusionnelle a été de 4,6 semaines. La durée médiane d’indépendance transfusionnelle n’a été atteinte dans aucun bras de traitement, mais elle devrait être supérieure à 2 ans chez les patients traités par le lénalidomide.
L’augmentation médiane du taux d’hémoglobine (Hb) depuis le début du traitement a été de 6,4 g/dL dans le bras 10 mg.
Les autres critères d’évaluation de l’étude étaient la réponse cytogénétique (dans le bras 10 mg, des réponses cytogénétiques majeures et mineures ont été observées chez respectivement 30,0 % et 24,0 % des patients), l’évaluation de la qualité de vie liée à la santé (QdVLS) et la progression en leucémie aiguë myéloblastique. Les résultats en termes de réponse cytogénétique et de qualité de vie ont concordé avec les résultats du critère principal et étaient en faveur du traitement par le lénalidomide par rapport au placebo.
Dans l’étude MDS-003, un pourcentage élevé (58,1 %) de patients atteints de syndromes myélodysplasiques traités par le lénalidomide 10 mg a atteint l’indépendance transfusionnelle (> 182 jours). Le délai médian jusqu’à l’indépendance transfusionnelle a été de 4,1 semaines. La durée médiane d’indépendance transfusionnelle a été de 114,4 semaines. L’augmentation médiane du taux d’hémoglobine (Hb) a été de 5,6 g/dL. Des réponses cytogénétiques majeures et mineures ont été observées chez 40,9 % et 30,7 % des patients respectivement.
Un pourcentage élevé de patients inclus dans les études MDS-003 (72,9 %) et MDS-004 (52,7 %) avaient reçu antérieurement des agents stimulant l’érythropoïèse.
· Lymphome à cellules du manteau
L’efficacité et la sécurité du lénalidomide ont été évaluées chez des patients présentant un lymphome à cellules du manteau dans une étude de phase 2 multicentrique, randomisée en ouvert, par rapport à une monothérapie choisie par l’investigateur, chez des patients qui étaient réfractaires à leur dernier traitement ou avaient rechuté une à trois fois (étude MCL-002).
Des patients âgés d’au moins 18 ans qui présentaient un lymphome à cellules du manteau confirmé par histologie et une maladie mesurable par TDM ont été inclus. Les patients devaient avoir reçu préalablement un traitement adéquat avec au moins un protocole de polychimiothérapie. De plus, les patients ne devaient pas être éligibles à une chimiothérapie intensive et/ou à une greffe au moment de l’inclusion dans l’étude. Les patients ont été randomisés selon un rapport 2:1 dans le bras lénalidomide ou dans le bras contrôle. Le traitement choisi par l’investigateur était sélectionné avant la randomisation et consistait en une monothérapie par chlorambucil, cytarabine, rituximab, fludarabine ou gemcitabine.
Le lénalidomide était administré par voie orale à la dose de 25 mg en une prise par jour pendant les 21 premiers jours (J1 à J21) de chaque cycle de 28 jours jusqu’à la progression de la maladie ou la survenue d’une toxicité inacceptable. Les patients présentant une insuffisance rénale modérée devaient recevoir une dose initiale plus faible de 10 mg de lénalidomide par jour selon le même schéma.
Les caractéristiques démographiques initiales étaient comparables entre le bras lénalidomide et le bras contrôle. L'âge médian était de 68,5 ans dans les deux populations de patients et le rapport hommes/femmes était comparable. L’indice de performance ECOG était comparable dans les deux groupes, ainsi que le nombre de traitements antérieurs.
Le critère principal d’évaluation de l’efficacité dans l’étude MCL-002 était la survie sans progression (SSP).
Les résultats d’efficacité dans la population en intention de traiter (ITT) étaient évalués par le Comité d’évaluation indépendant (IRC) et sont présentés dans le tableau ci-dessous.
Tableau 13. Synthèse des résultats d’efficacité – étude MCL-002, population en intention de traiter
|
|
Bras lénalidomide N = 170 |
Bras contrôle N = 84 |
|
SSP SSP, médianea [IC à 95 %]b (semaines) |
37,6 [24,0 ; 52,6] |
22,7 [15,9 ; 30,1] |
|
RR séquentiel [IC à 95 %]c |
0,61 [0,44 ; 0,84] |
|
|
Test du log-rank séquentiel, valeur pe |
0,004 |
|
|
Réponsea, n (%) |
|
|
|
Réponse complète (RC) |
8 (4,7) |
0 (0,0) |
|
Réponse partielle (RP) |
60 (35,3) |
9 (10,7) |
|
Maladie stable (MS)b |
50 (29,4) |
44 (52,4) |
|
Maladie en progression (MP) |
34 (20,0) |
26 (31,0) |
|
Non évaluée/Données manquantes |
18 (10,6) |
5 (6,0) |
|
TRG (RC, RCnc, RP), n (%) [IC à 95 %]c |
68 (40,0) [32,58 ; 47,78] |
9 (10,7)d [5,02 ; 19,37] |
|
Valeur pe |
< 0,001 |
|
|
Taux de RC (RC, RCnc), n (%) [IC à 95 %]c |
8 (4,7) [2,05 ; 9,06] |
0 (0,0) [95,70; 100,00] |
|
Valeur pe |
0,043 |
|
|
Durée de la réponse, médianea [IC à 95 %] (semaines) |
69,6 [41,1 ; 86,7] |
45,1 [36,3; 80,9] |
|
Survie globale |
|
|
|
RR [IC à 95 %]c |
0,89 [0,62 ; 1,28] |
|
|
Valeur p, test du log-rank |
0,520 |
|
IC = intervalle de confiance ; Taux de RC = taux de réponse complète ; RC = réponse complète ; RCnc = réponse complète non confirmée ; CSI = Comité de surveillance indépendant ; ITT = intention de traiter ; RR = rapport de risque ; KM = Kaplan-Meier ; MIPI = Mantle Cell Lymphoma International Prognostic Index ; S/O = sans objet ; TRG = taux de réponse globale ; MP = maladie en progression ; SSP = survie sans progression ; RP = réponse partielle ; GCS = greffe de cellules souches ; MS = maladie stable ; ES = erreur standard.
a La médiane était basée sur l’estimation de KM.
b L’intervalle était calculé comme les IC à 95 % de la durée de survie médiane.
c La moyenne et la médiane sont les statistiques univariées sans ajustement pour censure.
d Les variables de stratification étaient le délai entre le diagnostic et la première dose (< 3 ans et ≥ 3 ans), le délai entre le dernier traitement systémique antérieur du lymphome et la première dose (< 6 mois et ≥ 6 mois), les antécédents de GCS (oui ou non) et le score MIPI lors de l’inclusion (risque faible, intermédiaire et élevé).
e Le test séquentiel était basé sur une moyenne pondérée des statistiques d’un test du log-rank en utilisant le test du log-rank non stratifié pour l’augmentation de la taille de l’effectif et le test du log-rank non stratifié de l’analyse principale. Les pondérations sont basées sur les événements observés au moment de la troisième réunion du CSI et sur la différence entre les événements attendus et observés au moment de l’analyse principale. Le RR séquentiel associé et l’IC à 95 % correspondant sont présentés.
Dans la population ITT de l’étude MCL-002, il a été observé une augmentation apparente globale des décès au cours des 20 premières semaines dans le bras lénalidomide (22/170 patients [13 %]) par rapport au bras contrôle (6/84 patients [7 %]). Chez les patients ayant une charge tumorale élevée, les chiffres correspondants étaient de 16/81 patients (20 %) et 2/28 patients (7 %) (voir rubrique 4.4).
· Lymphome folliculaire
AUGMENT - CC-5013-NHL-007
L’efficacité et la sécurité du lénalidomide en association avec le rituximab versus rituximab plus placebo ont été évaluées chez des patients atteints d’un lymphome non hodgkinien indolent en rechute ou réfractaire, y compris un lymphome folliculaire, dans une étude de phase 3, multicentrique, randomisée, en double aveugle, contrôlée (CC-5013-NHL-007 [AUGMENT]).
Au total, 358 patients âgés de 18 ans et plus, atteints de LZM ou de LF de Grade 1, 2 ou 3a confirmé histologiquement (CD20+ par cytométrie en flux ou histochimie) tel qu’évalué par l’investigateur ou l’anatomo-pathologiste du centre, ont été randomisés selon un rapport 1:1. Les patients avaient reçu au moins un traitement antérieur par chimiothérapie systémique, immunothérapie ou immunochimiothérapie.
Le lénalidomide était administré par voie orale à la dose de 20 mg en une prise par jour pendant les 21 premiers jours de chaque cycle de 28 jours pendant 12 cycles ou jusqu’à la survenue d’une toxicité inacceptable. La dose de rituximab était de 375 mg/m2 chaque semaine au cours du cycle 1 (jours 1, 8, 15 et 22) et le jour 1 de chaque cycle de 28 jours pendant les cycles 2 à 5.
Toutes les doses de rituximab étaient calculées en fonction de la surface corporelle (SC) du patient, déterminée à partir du poids réel du patient.
Les caractéristiques démographiques et pathologiques initiales étaient comparables entre les deux groupes de traitement.
L’objectif principal de l’étude était de comparer l’efficacité du lénalidomide en association avec le rituximab à celle du rituximab plus placebo chez des patients atteints de LF de Grade 1, 2 ou 3a ou de LZM en rechute ou réfractaire.
Le critère principal d’évaluation de l’efficacité était la SSP, évaluée par un comité d’évaluation indépendant (IRC) selon les critères de réponse de l’International Working Group (CR-IWG) 2007, mais sans le recours à la tomographie par émission de positrons (TEP).
Les objectifs secondaires de l’étude étaient de comparer la tolérance du lénalidomide en association avec le rituximab à celle du rituximab plus placebo. Les autres objectifs secondaires étaient de comparer l’efficacité du rituximab plus lénalidomide à celle du rituximab plus placebo à l’aide des autres critères d’efficacité suivants : taux de réponse globale (RG), taux de RC et durée de la réponse (DR) selon les critères IWG 2007 sans TEP ainsi que la SG.
Les résultats dans la population totale incluant les patients atteints de LF ou de LZM ont montré qu’après un suivi médian de 28,3 mois, l’étude avait atteint son critère principal de SSP avec un rapport de risque (RR) (intervalle de confiance [IC]) à 95 % de 0,45 (0,33 ; 0,61), valeur p < 0,0001. Les résultats d’efficacité dans la population de patients atteints d’un lymphome folliculaire sont présentés dans le tableau 14.
Tableau 14. Synthèse des résultats d’efficacité chez les patients atteints d’un lymphome folliculaire – Étude CC-5013-NHL-007
|
|
LF (N = 295) |
|
|
|
Lénalidomide + Rituximab (N = 147) |
Placebo + Rituximab (N = 148) |
|
Survie sans progression (SSP) (données censurées selon les règles de l’EMA) |
||
|
SSP médianea, mois (IC à 95 %) |
39,4 |
13,8 |
|
(25,1, NE) |
(11,2, 16,0) |
|
|
RR [IC à 95 %] |
0,40 (0,29, 0,55)b |
|
|
Valeur p |
< 0,0001c |
|
|
Réponse objectived (RC +RP), n (%) (IRC-IWG 2007) IC à 95 %f |
118 (80,3) (72,9, 86,4) |
82 (55,4) (47,0, 63,6) |
|
Réponse complèted, n (%) |
51 (34,7) |
29 (19,6) |
|
(IRC-IWG 2007) |
||
|
|
(27,0, 43,0) |
(13,5, 26,9) |
|
IC à 95 %f |
||
|
Durée de la réponsed (médiane) (mois) IC à 95 %a |
36,6 (24,9, NE) |
15,5 (11,2, 25,0) |
|
Survie globaled,e (SG) |
||
|
Taux de SG à 5 ans, n (%) |
126 (85,9) |
114 (77,0) |
|
IC à 95 % |
(78,6, 90,9) |
(68,9, 83,3) |
|
RR [IC à 95 %] |
0,49 (0,28, 0,85)b |
|
|
Suivi |
|
|
|
Durée médiane du suivi, mois (min, max) |
67,81 |
65,72 |
|
|
(0,5, 50,9) |
(0,6, 50,9) |
ª Estimation de la médiane à partir de l’analyse de Kaplan-Meier
b Le rapport de risque et son intervalle de confiance ont été estimés à partir d’un modèle pour risques proportionnels de Cox non stratifié.
c Valeur p déterminée par le test log-rank
d Critères d’évaluation secondaires et exploratoires, sans contrôle du risque α.
e Après un suivi médian allant jusqu’à 66,14 mois, 1ç décès ont été observés dans le bras R2 et 38 dans le bras contrôle.
f Intervalle de confiance exact pour distribution binomiale.
· Lymphome folliculaire chez les patients réfractaires au rituximab
MAGNIFY - CC-5013-NHL-008
Au total, 232 patients âgés de 18 ans et plus, atteints de lymphome folliculaire (de Grade 1, 2 ou 3a ou lymphome de la zone marginale) confirmé histologiquement, tel qu’évalué par l’investigateur ou l’anatomo-pathologiste du centre, ont été inclus dans la période de traitement initiale comportant 12 cycles de traitement par le lénalidomide plus rituximab. Les patients ayant obtenu une RC/RC non confirmée (RCnc), une RP ou une MS à la fin de la période de traitement d’induction étaient randomisés pour entrer dans la période de traitement d’entretien. Tous les patients inclus devaient avoir reçu au moins un traitement systémique antérieur du lymphome. Contrairement à l’étude NHL-007, des patients réfractaires au rituximab (absence de réponse ou rechute dans les 6 mois suivant le traitement par le rituximab) ou réfractaires à la fois au rituximab et à la chimiothérapie ont été inclus dans l’étude NHL-008.
Pendant la période de traitement d’induction, le lénalidomide était administré à la dose de 20 mg les jours 1 à 21 de chaque cycle de 28 jours pendant 12 cycles au maximum ou jusqu’à la survenue d’une toxicité inacceptable, jusqu’au retrait du consentement ou jusqu’à la progression de la maladie. La dose de rituximab était de 375 mg/m2 chaque semaine au cours du cycle 1 (jours 1, 8, 15 et 22) puis le jour 1 d’un cycle sur deux de 28 jours (cycles 3, 5, 7, 9 et 11) pendant 12 cycles de traitement au maximum. Toutes les doses de rituximab étaient calculées en fonction de la surface corporelle (SC) du patient, déterminée à partir du poids réel du patient.
Les données présentées résultent d’une analyse intermédiaire portant sur la période de traitement d’induction en un seul bras. Les mesures de l’efficacité reposaient sur le taux de réponse globale (RG) par meilleure réponse utilisé comme critère principal d’évaluation, déterminé à l’aide des critères de réponse de l’International Working Group (CR-IWG) 1999. L’objectif secondaire de l’étude était d’évaluer les autres critères d’efficacité, tels que la durée de la réponse (DR).
Tableau 15. Synthèse des résultats d’efficacité (période de traitement d’induction) – Étude CC-5013-NHL-008
|
|
Tous les patients |
Patients atteints de LF |
||||
|
Total N = 187 a |
Réfractaires au rituximab: oui N = 77 |
Réfractaires au rituximab: non N = 110 |
Total N = 148 |
Réfractaires au rituximab: oui N = 60 |
Réfractaires au rituximab: non N = 88 |
|
|
Taux de réponse globale, n (%) (RC+RCnc+RP) |
127 (67,9) |
45 (58,4) |
82 (75,2) |
104 (70,3) |
35 (58,3) |
69 (79,3) |
|
Taux de réponse complète, n (%) (RC+RCnc) |
79 (42,2) |
27 (35,1) |
52 (47,7) |
62 (41,9) |
20 (33,3) |
42 (48,3) |
|
Nombre de répondeurs |
N = 127 |
N = 45 |
N = 82 |
N = 104 |
N = 35 |
N = 69 |
|
% de patients avec DRb |
93,0 |
90,4 |
94,5 |
94,3 |
96,0 |
93,5 |
|
≥ 6 mois (IC à 95 %)c |
(85,1, 96,8) |
(73,0, 96,8) |
(83,9, 98,2) |
(85,5, 97,9) |
(74,8, 99,4) |
(81,0, 97,9) |
|
% de patients avec DRb |
79,1 |
73,3 |
82,4 |
79,5 |
73,9 |
81,7 |
|
≥ 12 mois (IC à 95 %)c |
(67,4, 87,0) |
(51,2, 86,6) |
(67,5, 90,9) |
(65,5, 88,3) |
(43,0, 89,8) |
(64,8, 91,0) |
IC = intervalle de confiance ; DR = durée de la réponse ; LF = lymphome folliculaire
a La population principale d’analyse est la population évaluable pour l’efficacité de l’induction (EEI).
b La durée de réponse est définie comme le temps (mois) écoulé entre la réponse initiale (au moins un RP) et la progression documentée de la maladie ou le décès, selon l’événement survenant en premier.
c Les données statistiques ont été obtenues selon la méthode de Kaplan-Meier, l’IC à 95 % a été déterminé selon la méthode de Greenwood.
Remarques : l’analyse a été effectuée uniquement chez les patients ayant obtenu au moins une RP après la date de première administration du traitement d’induction et avant tout traitement de la période d’entretien et tout traitement ultérieur du lymphome pendant la période d’induction. Le pourcentage a été calculé en fonction du nombre total de répondeurs.
Population pédiatrique
L’Agence européenne des médicaments (EMA) a accordé une dérogation spécifique au produit de référence qui s’applique à tous les sous-groupes de la population pédiatrique dans les indications de tumeurs à cellules B matures (voir rubrique 4.2 pour les informations concernant l’usage pédiatrique).
5.2. Propriétés pharmacocinétiques
Absorption
Le lénalidomide est rapidement absorbé après administration orale chez les volontaires sains à jeun ; les concentrations plasmatiques maximales sont atteintes entre 0,5 et 2 heures après la prise. Chez les patients, ainsi que chez les volontaires sains, la concentration maximale (Cmax) et l’aire sous la courbe de concentration en fonction du temps (ASC) augmentent proportionnellement à l’augmentation de la dose. En cas de prises multiples aucune accumulation notable du médicament n’est notée. Dans le plasma, la disponibilité systémique relative approximative des énantiomères R- et S- est respectivement de 56 % et 44 %.
La co-administration avec un repas hyperlipidique et hypercalorique chez les volontaires sains, diminue la quantité absorbée, ce qui entraîne une diminution d’environ 20 % de l’aire sous la courbe de concentration en fonction du temps (ASC) et une réduction de 50 % de la Cmax plasmatique. Cependant, dans les principales études d’enregistrement menées dans le myélome multiple et les syndromes myélodysplasiques ayant permis d’établir l’efficacité et la sécurité du lénalidomide, le médicament a été administré indépendamment de la prise d’aliments. Le lénalidomide peut donc être administré pendant ou en dehors des repas.
Les analyses pharmacocinétiques de population indiquent que le taux d’absorption du lénalidomide administré par voie orale est comparable chez les patients présentant un myélome multiple, un syndrome myélodysplasique ou un lymphome à cellules du manteau.
Distribution
In vitro, le lénalidomide marqué (14C) s’est faiblement lié aux protéines plasmatiques, le taux de liaison moyen avec les protéines plasmatiques ayant été respectivement de 23 % et 29 % chez les patients atteints de myélomes multiples et chez les volontaires sains.
Le lénalidomide est présent dans le sperme humain (< 0,01 % de la dose) après administration de 25 mg/jour et il est indétectable dans le sperme d’un sujet sain trois jours après l’arrêt du médicament (voir rubrique 4.4).
Biotransformation et élimination
Les résultats des études in vitro du métabolisme humain indiquent que le lénalidomide n’est pas métabolisé par les enzymes du cytochrome P450, ce qui permet de penser que l’administration de lénalidomide avec des médicaments inhibant les enzymes du cytochrome P450 ne devrait pas entraîner d’interactions médicamenteuses métaboliques chez l’homme. Les études in vitro indiquent que le lénalidomide n’a pas d’effet inhibiteur sur les enzymes CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A ou UGT1A1. Par conséquent, il est peu probable que le lénalidomide provoque des interactions cliniquement significatives en cas d’administration concomitante avec des substrats de ces enzymes.
Les études in vitro indiquent que le lénalidomide n’est pas un substrat de la protéine BCRP (Breast cancer resistance protein) humaine, des transporteurs de la famille MRP (multidrug resistance protein) MRP1, MRP2 ou MRP3, des transporteurs d’anions organiques (OAT) OAT1 et OAT3, du polypeptide 1B1 de transport des anions organiques (OATP1B1), des transporteurs de cations organiques (OCT) OCT1 et OCT2, de la protéine MATE1 de la famille multidrug and toxin extrusion protein (MATE) et des nouveaux types de transporteurs de cations organiques (OCTN – organic cation transporters novel) OCTN1 et OCTN2.
Les études in vitro indiquent que le lénalidomide n’a pas d’effet inhibiteur sur les protéines humaines BSEP (bile salt export pump – pompe d’exportation des sels biliaires), BCRP, MRP2, OAT1, OAT3, OATP1B1, OATP1B3 et OCT2.
La majorité du lénalidomide est éliminée par excrétion rénale. La contribution de l’excrétion rénale à la clairance totale chez un sujet à la fonction rénale normale était de 90 % avec 4 % du lénalidomide éliminé dans les fèces.
Le lénalidomide est peu métabolisé car 82 % de la dose sont excrétés sous forme inchangée dans l’urine. L’hydroxy-lénalidomide et le N-acétyl-lénalidomide représentent respectivement 4,59 % et 1,83 % de la dose excrétée. La clairance rénale du lénalidomide est supérieure au taux de filtration glomérulaire et est donc, au moins, activement secrété dans une certaine mesure.
Aux doses de 5 à 25 mg/jour, la demi-vie plasmatique est d’environ 3 heures chez les volontaires sains et de 3 à 5 heures chez les patients atteints de myélome multiple, de syndromes myélodysplasiques ou de lymphome à cellules du manteau.
Sujets âgés
Il n’a pas été mené d’études cliniques spécifiques pour évaluer la pharmacocinétique du lénalidomide chez les sujets âgés. Les analyses pharmacocinétiques de population incluaient des patients âgés de 39 à 85 ans et indiquent que l'âge n’a pas d’effet sur l’élimination du lénalidomide (exposition plasmatique). Puisque les patients âgés sont plus susceptibles d’avoir une diminution de la fonction rénale, le choix de la posologie devra être fait avec précaution chez ces patients et il est recommandé de surveiller leur fonction rénale.
Insuffisance rénale
La pharmacocinétique du lénalidomide a été étudiée chez des sujets présentant une insuffisance rénale due à des pathologies non malignes. Dans cette étude, deux méthodes ont été utilisées pour classifier la fonction rénale : la clairance de la créatinine urinaire mesurée sur 24 heures et la clairance de la créatinine estimée selon la formule de Cockcroft-Gault. Les résultats indiquent que quand la fonction rénale diminue (< 50 mL/min), la clairance totale du lénalidomide décroît proportionnellement entrainant une augmentation de l’ASC. L’ASC a été multipliée par environ 2,5, 4 et 5 chez les sujets présentant une insuffisance rénale modérée, une insuffisance rénale sévère et une insuffisance rénale terminale, respectivement, par rapport au groupe combinant les sujets ayant une fonction rénale normale et une insuffisance rénale légère. La demi-vie du lénalidomide a augmenté de 3,5 heures environ chez les patients avec une clairance de la créatinine > 50 mL/min à plus de 9 heures chez les patients avec une diminution de la fonction rénale < 50 mL/min. Cependant, l’insuffisance rénale n’a pas eu d’incidence sur l’absorption orale du lénalidomide. La Cmax a été similaire chez les sujets sains et chez les patients présentant une insuffisance rénale. Environ 30 % du médicament absorbé ont été éliminés au cours d’une séance de dialyse unique de 4 heures. Les ajustements posologiques recommandés en cas d’insuffisance rénale sont décrits dans la rubrique 4.2.
Insuffisance hépatique
Les analyses pharmacocinétiques de population incluaient des patients présentant une insuffisance hépatique légère (N = 16, bilirubine totale > 1 et ≤ 1,5 x LSN ou ASAT > LSN) ; et indiquent que l’insuffisance hépatique légère n’a pas d’effet sur l’élimination du lénalidomide (exposition plasmatique). Il n’existe pas de données chez les patients atteints d’insuffisance hépatique modérée à sévère.
Autres facteurs intrinsèques
Les analyses pharmacocinétiques de population indiquent que le poids (33 à 135 kg), le sexe, la race et le type d’hémopathie maligne (myélome multiple, syndrome myélodysplasique ou lymphome à cellules du manteau) n’ont pas d’effet cliniquement pertinent sur l’élimination du lénalidomide chez les patients adultes.
5.3. Données de sécurité préclinique
Plusieurs effets viscéraux (décoloration, foyers rouges sur différents organes, petite masse incolore au-dessus de la valvule auriculo-ventriculaire, hypotrophie de la vésicule biliaire, malformations du diaphragme) ont également été observés chez des fœtus isolés.
Le lénalidomide peut potentiellement présenter un risque de toxicité aiguë, la dose létale minimale par voie orale a été de > 2 000 mg/kg/jour chez les rongeurs. Chez le rat, l’administration orale répétée de 75, 150 et 300 mg/kg/jour pendant une durée allant jusqu’à 26 semaines a engendré une amplification réversible, liée au traitement, de la calcification du calice rénal aux 3 doses, plus particulièrement chez les femelles. La dose maximale sans effet indésirable observé (no observed adverse effect level, NOAEL) a été estimée à moins de 75 mg/kg/jour et est environ 25 fois supérieure à l’exposition quotidienne telle que définie par l’ASC, chez l’être humain. Chez le singe l’administration orale répétée de 4 et 6 mg/kg/jour pendant une durée allant jusqu’à 20 semaines a engendré une mortalité et une toxicité significative (perte de poids marquée, diminution du nombre des érythrocytes, des leucocytes et de la numération plaquettaire, hémorragie organique multiple, inflammation du tractus gastro-intestinal, atrophie lymphoïde et de la moelle osseuse). Chez le singe, l’administration orale répétée de 1 et 2 mg/kg/jour pendant une durée allant jusqu’à 1 an a engendré des modifications réversibles de la cellularité de la moelle osseuse, une légère réduction du rapport cellules myéloïdes/érythroïdes et une atrophie thymique. Une légère réduction de la numération leucocytaire a été notée à la dose de 1 mg/kg/jour correspondant approximativement à la même dose chez l’être humain, en se basant sur les comparaisons des ASC.
Les études de la mutagénicité in vitro (mutation bactérienne, lymphocytes humains, lymphomes de souris, transformation de cellules embryonnaires de hamster syrien) et in vivo (test des micronoyaux chez le rat) n’ont mis en évidence aucun effet lié au médicament, que ce soit au niveau génétique ou chromosomique. Aucune étude n’a été réalisée concernant la cancérogénicité du lénalidomide.
Des études de la toxicité sur le développement ont été précédemment menées chez le lapin. Dans ces études, les lapins ont reçu des doses orales de 3, 10 et 20 mg/kg/jour. Une absence du lobe intermédiaire du poumon a été observée à 10 et 20 mg/kg/jour, en rapport avec la dose, et une ectopie rénale a été observée à 20 mg/kg/jour. Bien que ces effets aient été notés à des doses toxiques pour la mère, ils pourraient être le résultat d’un effet direct. Des altérations des tissus mous et du squelette chez le fœtus ont également été observées aux doses de 10 et 20 mg/kg/jour.
Lactose
Cellulose microcristalline (E460(i))
Croscarmellose sodique (E468)
Stéarate de magnésium (E470b)
Enveloppe de la gélule
Gélatine
Dioxyde de titane (E171)
Indigotine (E132)
Oxyde de fer jaune (E172)
Encre d’impression
Gomme laque (E904)
Propylène glycol (E1520)
Oxyde de fer noir (E172)
Hydroxyde de potassium (E525)
3 ans.
6.4. Précautions particulières de conservation
Ce médicament ne nécessite pas de précautions particulières de conservation.
6.5. Nature et contenu de l'emballage extérieur
Plaquettes (OPA/Aluminium/PVC/Aluminium).
Boîte de 7, 14, 21, 28 et 42 gélules.
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
Les gélules ne doivent pas être ouvertes ou écrasées. Si la poudre de lénalidomide entre en contact avec la peau, laver immédiatement et abondamment la peau au savon et à l’eau. En cas de contact du lénalidomide avec les muqueuses, rincer abondamment à l’eau.
Les professionnels de santé et les aidants doivent porter des gants jetables pour manipuler la plaquette ou la gélule.
Les gants doivent ensuite être retirés avec précaution afin d’éviter une exposition cutanée, placés dans un sac plastique en polyéthylène à fermeture hermétique et éliminés conformément à la réglementation en vigueur. Les mains doivent ensuite être soigneusement lavées au savon et à l’eau. Les femmes enceintes ou qui pensent l’être ne doivent pas manipuler la plaquette ou la gélule (voir rubrique 4.4).
Tout médicament non utilisé ou déchet doit être retourné à un pharmacien pour une élimination en toute sécurité conformément à la réglementation en vigueur.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
30 RUE EDOUARD NIEUPORT
69008 LYON
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 302 049 5 5 : 21 gélules sous plaquettes (OPA/Aluminium/PVC/Aluminium).
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[À compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[À compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I
Médicament soumis à prescription hospitalière.
Prescription réservée aux spécialistes en oncologie ou en hématologie, ou aux médecins compétents en cancérologie ou en maladies du sang.
Médicament nécessitant une surveillance particulière pendant le traitement :
· Pour tous les patients : la prescription nécessite la signature de l'accord de soins.
· Pour les femmes susceptibles de procréer :
o La prescription est limitée à 1 mois de traitement ;
o Un test de grossesse doit être réalisé tous les mois, dans les 3 jours précédant la prescription ; la date et le résultat du test de grossesse doivent être mentionnés dans le carnet patient ;
o La délivrance doit être effectuée au plus tard 7 jours après la prescription et après avoir vérifié la date et le résultat du test de grossesse.
ANSM - Mis à jour le : 26/07/2024
LENALIDOMIDE STRAGEN 10 mg, gélule
lénalidomide
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin ou votre pharmacien.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que LENALIDOMIDE STRAGEN 10 mg, gélule, et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant de prendre LENALIDOMIDE STRAGEN 10 mg, gélule ?
3. Comment prendre LENALIDOMIDE STRAGEN 10 mg, gélule ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver LENALIDOMIDE STRAGEN 10 mg, gélule ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE LENALIDOMIDE STRAGEN 10 mg, gélule ET DANS QUELS CAS EST-IL UTILISE ?
Classe pharmacothérapeutique : IMMUNOSUPPRESSEURS, AUTRES IMMUNOSUPPRESSEURS - Code ATC : L04AX04.
Qu’est-ce que LENALIDOMIDE STRAGEN 10 mg, gélule
LENALIDOMIDE STRAGEN 10 mg, gélule contient la substance active « lénalidomide ». Ce médicament appartient à un groupe de médicaments qui modifient le fonctionnement de votre système immunitaire.
Dans quels cas LENALIDOMIDE STRAGEN 10 mg, gélule
LENALIDOMIDE STRAGEN 10 mg, gélule est utilisé chez les patients adultes dans le traitement :
· du myélome multiple
· des syndromes myélodysplasiques
· du lymphome à cellules du manteau
· du lymphome folliculaire
Myélome multiple
Le myélome multiple est un type de cancer touchant un certain type de cellules sanguines appelées plasmocytes. Ces cellules s’accumulent dans la moelle osseuse et se multiplient et deviennent incontrôlées. Cela peut entraîner une atteinte des os et des reins.
En général, le myélome multiple ne peut pas être guéri. Cependant, les signes et symptômes peuvent régresser de façon importante ou disparaître pendant une certaine période. Cela est appelé une « rémission ».
Myélome multiple non préalablement traité - chez les patients qui ont reçu une greffe de moelle osseuse
Dans cette indication, LENALIDOMIDE STRAGEN 10 mg, gélule est utilisé Dans cette indication, traitement d’entretien après une récupération adéquate des patients à la suite d’une greffe de moelle osseuse.
Myélome multiple non préalablement traité – chez les patients qui ne peuvent pas être traités par une greffe de moelle osseuse
LENALIDOMIDE STRAGEN 10 mg, gélule est pris avec d’autres médicaments. Ceux-ci peuvent être :
· un médicament de chimiothérapie appelé « bortézomib »
· un médicament anti-inflammatoire appelé « dexaméthasone »
· un médicament de chimiothérapie appelé « melphalan » et
· un médicament anti-inflammatoire appelé « prednisone »
Vous prendrez ces autres médicaments au début du traitement et vous continuerez ensuite en prenant LENALIDOMIDE STRAGEN 10 mg, gélule seul.
Si vous êtes âgé(e) de 75 ans ou plus ou si vous présentez des troubles rénaux modérés à sévères, votre médecin effectuera une évaluation attentive avant le début du traitement.
Myélome multiple - chez les patients qui ont déjà été traités
LENALIDOMIDE STRAGEN 10 mg, gélule est pris en association avec un médicament anti-inflammatoire appelé « dexaméthasone ».
LENALIDOMIDE STRAGEN 10 mg, gélule peut empêcher l’aggravation des signes et symptômes du myélome multiple. Il a également été démontré qu’il retarde la récidive du myélome multiple après le traitement.
Syndromes myélodysplasiques (SMD)
Le terme SMD désigne un ensemble de nombreuses maladies différentes du sang et de la moelle osseuse. Les cellules sanguines deviennent anormales et ne fonctionnent pas correctement. Les patients peuvent présenter différents signes et symptômes, notamment un taux faible de globules rouges (anémie), un besoin de transfusions sanguines et un risque d’infection.
LENALIDOMIDE STRAGEN 10 mg, gélule est utilisé seul pour traiter les patients adultes chez lesquels un SMD a été diagnostiqué et lorsque toutes les conditions ci-dessous sont remplies :
· vous avez besoin de transfusions sanguines régulières pour corriger un taux faible de globules rouges (« anémie avec dépendance transfusionnelle »)
· vous présentez une anomalie des cellules dans la moelle osseuse appelée « anomalie cytogénétique de délétion 5q isolée ». Cela signifie que votre organisme ne fabrique pas assez de cellules sanguines saines
· d’autres traitements qui ont été administrés préalablement, ne sont pas adaptés ou ne sont pas suffisamment efficaces
LENALIDOMIDE STRAGEN 10 mg, gélule peut augmenter le nombre de globules rouges normaux produits par l’organisme en diminuant le nombre de cellules anormales :
· Cela peut réduire le nombre de transfusions sanguines nécessaires. Il est possible que le recours aux transfusions ne soit plus nécessaire.
Lymphome à cellules du manteau (LCM)
Le LCM est un cancer d‘une partie du système immunitaire (le tissu lymphoïde). Il touche un type de globules blancs appelés lymphocytes B ou cellules B. Le LCM est une maladie dans laquelle les lymphocytes B se multiplient de façon incontrôlée et s’accumulent dans le tissu lymphoïde, la moelle osseuse ou le sang.
LENALIDOMIDE STRAGEN 10 mg, gélule est utilisé seul pour traiter les patients adultes qui ont été préalablement traités avec d’autres médicaments.
Lymphome folliculaire (LF)
Le LF est un cancer à progression lente qui touche les lymphocytes B, un type de globules blancs qui aident votre organisme à lutter contre les infections. En cas de LF, un nombre excessif de ces lymphocytes B peut s’accumuler dans le sang, la moelle osseuse, les ganglions lymphatiques et la rate.
LENALIDOMIDE STRAGEN 10 mg, gélule est pris en association avec un autre médicament appelé « rituximab » pour le traitement des patients adultes atteints de lymphome folliculaire préalablement traité.
Comment agit LENALIDOMIDE STRAGEN 10 mg, gélule ?
LENALIDOMIDE STRAGEN 10 mg, gélule agit en modifiant le fonctionnement du système immunitaire de l’organisme et en attaquant directement le cancer. Il agit de plusieurs façons différentes :
· en arrêtant le développement des cellules cancéreuses,
· en arrêtant la croissance des vaisseaux sanguins dans la tumeur,
· en stimulant une partie du système immunitaire pour attaquer les cellules cancéreuses.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT DE PRENDRE LENALIDOMIDE STRAGEN 10 mg, gélule ?
Ne prenez jamais LENALIDOMIDE STRAGEN 10 mg, gélule :
· Si vous êtes enceinte, si vous pensez que vous pourriez l’être ou si vous prévoyez de l'être, car un effet nocif du LENALIDOMIDE STRAGEN 10 mg, gélule est attendu pour l’enfant à naître (voir la section « Grossesse, allaitement et contraception - Informations pour les femmes et les hommes » de la rubrique 2),
· Si vous êtes en âge de devenir enceinte, sauf si vous prenez toutes les mesures nécessaires pour ne pas être enceinte (voir la section « Grossesse, allaitement et contraception - Informations pour les femmes et les hommes » de la rubrique 2). Si vous êtes en âge de devenir enceinte, votre médecin notera à chaque prescription que les mesures nécessaires ont été prises et vous en donnera confirmation,
· Si vous êtes allergique au lénalidomide ou à l’un des autres composants contenus dans ce médicament mentionnés dans la rubrique 6. Si vous pensez que vous pourriez être allergique, demandez conseil à votre médecin.
Si l’une de ces situations vous concerne, ne prenez pas LENALIDOMIDE STRAGEN 10 mg, gélule. Adressez-vous à votre médecin en cas de doute.
Avertissements et précautions
Adressez-vous à votre médecin, à votre pharmacien ou à votre infirmier/ère avant de prendre LENALIDOMIDE STRAGEN 10 mg, gélule si :
· Vous avez des antécédents de caillots sanguins. Vous avez un risque plus important de formation de caillots sanguins dans les veines et les artères pendant le traitement,
· Vous présentez des signes d’infection tels que toux ou fièvre,
· Vous avez ou avez eu dans le passé une infection virale, en particulier hépatite B, varicelle, zona ou infection par le VIH. En cas de doute, adressez-vous à votre médecin. Le traitement par LENALIDOMIDE STRAGEN 10 mg, gélule peut provoquer une réactivation du virus chez les patients qui en sont porteurs. Cela entraîne une récidive de l’infection. Votre médecin doit déterminer si vous avez eu dans le passé une hépatite B.
· Vous avez des problèmes de reins ; votre médecin pourra adapter votre dose de LENALIDOMIDE STRAGEN 10 mg, gélule,
· Vous avez des antécédents de crise cardiaque (infarctus du myocarde), vous avez déjà développé un caillot sanguin ou si vous fumez, vous avez une pression artérielle élevée (hypertension) ou un taux de cholestérol élevé,
· Vous avez développé une réaction allergique pendant un traitement par le thalidomide (un autre médicament utilisé dans le traitement du myélome multiple), par exemple éruption cutanée, démangeaisons, gonflement (œdème), vertiges ou difficultés à respirer.
· Vous avez déjà présenté simultanément plusieurs des symptômes suivants : éruption cutanée étendue, rougeurs cutanées, température élevée, symptômes ressemblant à ceux de la grippe, augmentation des enzymes hépatiques, anomalies sanguines (éosinophilie), augmentation du volume des ganglions lymphatiques – ce sont des signes de réaction cutanée sévère appelée réaction médicamenteuse accompagnée d’une éosinophilie et de symptômes systémiques, appelée également syndrome DRESS ou syndrome d’hypersensibilité médicamenteuse (voir également la rubrique 4 « Quels sont les effets indésirables éventuels ? »).
Si l’une de ces situations vous concerne, adressez-vous à votre médecin, pharmacien ou infirmier/ère avant de commencer le traitement.
À tout moment pendant ou après votre traitement, informez immédiatement votre médecin ou votre infirmier/ère si vous :
· avez une perte de vision, vous voyez floue ou trouble, des difficultés à parler, une faiblesse à un bras ou une jambe, un changement dans la façon dont vous marchez ou des problèmes d'équilibre, un engourdissement persistant, une diminution de sensation ou une perte de sensation, une perte de mémoire ou une confusion. Il peut s'agir de symptômes d'une affection cérébrale grave et potentiellement mortelle connue sous le nom de leucoencéphalopathie multifocale progressive (LEMP). Si vous aviez ces symptômes avant le traitement au lénalidomide, informez votre médecin de tout changement dans ces symptômes.
Examens et contrôles
Avant et pendant le traitement par LENALIDOMIDE STRAGEN 10 mg, gélule vous ferez régulièrement des analyses de sang car LENALIDOMIDE STRAGEN 10 mg, gélule peut faire chuter le nombre de cellules sanguines qui contribuent à lutter contre les infections (globules blancs) et de cellules qui font coaguler le sang (plaquettes). Votre médecin vous demandera de faire réaliser une analyse de sang :
· Avant le traitement
· Chaque semaine pendant les 8 premières semaines de traitement
· Puis au moins une fois par mois par la suite
Pour les patients présentant un syndrome myélodysplasique (SMD) et prenant LENALIDOMIDE STRAGEN 10 mg, gélule
Si vous présentez un SMD, vous pouvez avoir un risque plus élevé de développer une maladie plus avancée appelée leucémie aiguë myéloblastique (LAM). En outre, l’effet de LENALIDOMIDE STRAGEN sur le risque de développer une LAM n’est pas connu. Votre médecin pourra donc demander des examens afin de rechercher des signes permettant de mieux prédire la probabilité que vous développiez une LAM pendant votre traitement par LENALIDOMIDE STRAGEN.
Pour les patients présentant un lymphome à cellules du manteau (LCM) et prenant LENALIDOMIDE STRAGEN 10 mg, gélule
Votre médecin vous demandera de faire réaliser une analyse de sang :
· Avant le traitement
· Chaque semaine pendant les 8 premières semaines de traitement (2 cycles)
· Puis toutes les 2 semaines pendant les cycles 3 et 4 (voir rubrique 3 « Cycle de traitement » pour des informations plus détaillées)
· L’analyse de sang sera ensuite effectuée au début de chaque cycle et
· Au minimum tous les mois
Pour les patients présentant un lymphome folliculaire (LF) et prenant LENALIDOMIDE STRAGEN 10 mg, gélule
Votre médecin vous demandera de faire réaliser une analyse de sang :
· Avant le traitement
· Chaque semaine pendant les 3 premières semaines de traitement (1 cycle)
· Puis toutes les 2 semaines pendant les cycles 2 à 4 (voir rubrique 3 « Cycle de traitement » pour des informations plus détaillées)
· L’analyse de sang sera ensuite effectuée au début de chaque cycle et
· Au minimum tous les mois
Votre médecin pourra demander un examen afin de déterminer si vous avez une charge tumorale totale élevée dans tout le corps, y compris dans la moelle osseuse. Cela peut provoquer une maladie dans laquelle la destruction des cellules cancéreuses entraîne des taux anormaux de substances chimiques dans le sang, ce qui peut provoquer une insuffisance rénale (cette maladie est appelée « syndrome de lyse tumorale »).
Votre médecin pourra également vous examiner pour détecter des modifications de votre peau telles que des taches rouges ou des éruptions cutanées.
Votre médecin peut décider d’ajuster la dose de LENALIDOMIDE STRAGEN 10 mg, gélule que vous prenez ou d’arrêter le traitement en fonction des résultats des analyses de sang et de votre état général. Si vous présentez un myélome multiple récemment diagnostiqué, votre médecin pourra également évaluer votre traitement en fonction de votre âge et des autres maladies que vous présentez déjà.
Dons de sang
Vous ne devez pas faire des dons de sang pendant le traitement et pendant au moins 7 jours après la fin du traitement.
Enfants et adolescents
L’utilisation de LENALIDOMIDE STRAGEN 10 mg, gélule n’est pas recommandée chez les enfants et adolescents de moins de 18 ans.
Patients âgés et patients ayant des troubles rénaux
Si vous êtes âgé(e) de 75 ans ou plus ou que vous avez des troubles rénaux modérés à sévères, votre médecin effectuera une évaluation attentive avant le début du traitement.
Autres médicaments et LENALIDOMIDE STRAGEN 10 mg, gélule
Informez votre médecin ou pharmacien si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament, car LENALIDOMIDE STRAGEN 10 mg, gélule peut modifier la façon dont certains médicaments agissent. De même, certains autres médicaments peuvent modifier la façon dont LENALIDOMIDE STRAGEN 10 mg, gélule agit.
En particulier, prévenez votre médecin ou votre infirmier/ère si vous prenez l’un des médicaments suivants :
· Certains médicaments utilisés pour éviter une grossesse tels que les contraceptifs oraux, car ils pourraient ne plus être efficaces
· Certains médicaments utilisés pour traiter les problèmes cardiaques, tels que la digoxine
· Certains médicaments utilisés pour fluidifier le sang, tels que la warfarine
Grossesse, allaitement et contraception – Informations pour les femmes et les hommes
Grossesse
Pour les femmes prenant LENALIDOMIDE STRAGEN 10 mg, gélule
· Vous ne devez pas prendre LENALIDOMIDE STRAGEN 10 mg, gélule si vous êtes enceinte, car un effet nocif de ce médicament est attendu pour l’enfant à naître.
· Vous ne devez pas devenir enceinte pendant que vous prenez LENALIDOMIDE STRAGEN 10 mg, gélule. Vous devez donc utiliser un moyen de contraception efficace si vous êtes une femme en âge de devenir enceinte (voir la section « Contraception »).
· En cas de grossesse pendant votre traitement par LENALIDOMIDE STRAGEN 10 mg, gélule vous devez arrêter le traitement et informer immédiatement votre médecin.
Pour les hommes prenant LENALIDOMIDE STRAGEN 10 mg, gélule
· Si votre partenaire devient enceinte pendant que vous prenez LENALIDOMIDE STRAGEN 10 mg, gélule, vous devez informer votre médecin immédiatement. Il est recommandé que votre partenaire consulte un médecin.
· Vous devez également utiliser un moyen de contraception efficace (voir la section « Contraception »).
Allaitement
Vous ne devez pas allaiter pendant que vous prenez LENALIDOMIDE STRAGEN 10 mg, gélule car on ignore si LENALIDOMIDE STRAGEN 10 mg, gélule passe dans le lait maternel.
Contraception
Pour les femmes prenant LENALIDOMIDE STRAGEN 10 mg, gélule
Avant de débuter le traitement, vérifiez auprès de votre médecin si vous êtes en âge de devenir enceinte, même si cela vous semble improbable.
Si vous êtes en âge de devenir enceinte :
· Vous devrez réaliser un test de grossesse sous le contrôle de votre médecin (avant le traitement, au moins toutes les 4 semaines pendant le traitement et au moins 4 semaines après la fin du traitement) sauf s’il est confirmé que les trompes de Fallope ont été sectionnées et oblitérées afin d’empêcher les ovules d’arriver dans l’utérus (stérilisation tubaire),
ET
· Vous devrez utiliser une méthode de contraception efficace. Cette contraception doit être débutée au moins 4 semaines avant le début du traitement, poursuivie pendant toute la durée du traitement et durant au moins 4 semaines après l’arrêt du traitement. Votre médecin vous conseillera sur les moyens de contraception adaptés à votre cas.
Pour les hommes prenant LENALIDOMIDE STRAGEN 10 mg, gélule
LENALIDOMIDE STRAGEN 10 mg, gélule passe dans le sperme humain. Si votre partenaire est enceinte ou susceptible de le devenir et qu’elle n’utilise pas un moyen de contraception efficace, vous devez utiliser des préservatifs pendant le traitement et pendant au moins 7 jours après la fin du traitement, même si vous avez subi une vasectomie. Vous ne devez pas faire des dons de sperme pendant le traitement et pendant au moins 7 jours après la fin du traitement.
Conduite de véhicules et utilisation de machines
Il est déconseillé de conduire ou d’utiliser des machines si vous ressentez des étourdissements, une fatigue, de la somnolence, des vertiges ou si votre vision est trouble après avoir pris LENALIDOMIDE STRAGEN 10 mg, gélule.
LENALIDOMIDE STRAGEN 10 mg, gélule contient du lactose
Si votre médecin vous a informé(e) d’une intolérance à certains sucres, contactez-le avant de prendre ce médicament.
LENALIDOMIDE STRAGEN 10 mg, gélule contient du sodium
Ce médicament contient moins de 1 mmol (23 mg) de sodium c‘est-à-dire qu’il est essentiellement « sans sodium ».
3. COMMENT PRENDRE LENALIDOMIDE STRAGEN 10 mg, gélule ?
· Dans le traitement des patients atteints de myélome multiple qui ne peuvent pas recevoir de greffe de moelle osseuse ou qui ont déjà reçu d’autres traitements auparavant, LENALIDOMIDE STRAGEN 10 mg, gélule est utilisé avec d’autres médicaments (voir la rubrique 1 « Qu’est-ce que LENALIDOMIDE STRAGEN 10 mg, gélule et dans quel cas est-il utilisé ? »).
· Dans le traitement des patients atteints de myélome multiple qui ont reçu une greffe de moelle osseuse ou des patients atteints de SMD ou de LCM, LENALIDOMIDE STRAGEN 10 mg, gélule est utilisé seul.
· Dans le traitement des patients atteints de lymphome folliculaire, LENALIDOMIDE STRAGEN 10 mg, gélule est utilisé avec un autre médicament appelé « rituximab ».
Veillez toujours à prendre ce médicament en suivant exactement les indications de votre médecin. Vérifiez auprès de votre médecin ou pharmacien en cas de doute.
Si vous prenez LENALIDOMIDE STRAGEN 10 mg, gélule en association avec d’autres médicaments, reportez-vous aux notices de ces médicaments pour plus d’informations sur leur utilisation et leurs effets.
Cycle de traitement
LENALIDOMIDE STRAGEN 10 mg, gélule est pris certains jours de périodes de 3 semaines (21 jours).
· Chaque période de 21 jours est appelée un « cycle de traitement »
· Selon le jour du cycle, vous prendrez un ou plusieurs médicaments. Certains jours toutefois, vous ne prendrez aucun des médicaments
· Après avoir terminé chaque cycle de 21 jours, vous devrez commencer un nouveau « cycle » pendant les 21 jours suivants
OU
LENALIDOMIDE STRAGEN 10 mg, gélule est pris certains jours de périodes de 4 semaines (28 jours).
· Chaque période de 28 jours est appelée un « cycle de traitement »
· Selon le jour du cycle, vous prendrez un ou plusieurs médicaments. Certains jours toutefois, vous ne prendrez aucun des médicaments
· Après avoir terminé chaque cycle de 28 jours, vous devrez commencer un nouveau « cycle » pendant les 28 jours suivants
Quelle dose de LENALIDOMIDE STRAGEN 10 mg, gélule prendre ?
Avant le début du traitement, votre médecin vous dira :
· Quelle dose de LENALIDOMIDE STRAGEN 10 mg, gélule vous devez prendre,
· Quelle dose des autres médicaments vous devez prendre en association avec LENALIDOMIDE STRAGEN 10 mg, gélule le cas échéant,
· Quels jours de votre cycle de traitement vous devez prendre chaque médicament.
Comment et quand prendre LENALIDOMIDE STRAGEN 10 mg, gélule ?
· Vous devez avaler les gélules entières, de préférence avec de l’eau.
· N’ouvrez pas les gélules, ne les cassez pas et ne les mâchez pas. Si la poudre d’une gélule de LENALIDOMIDE STRAGEN 10 mg, gélule ouverte entre en contact avec la peau, lavez immédiatement et abondamment la peau au savon et à l’eau.
· Les professionnels de santé, les aidants et les membres de la famille doivent porter des gants jetables pour manipuler la plaquette ou la gélule. Les gants doivent ensuite être retirés avec précaution afin d’éviter une exposition cutanée, placés dans un sac plastique en polyéthylène à fermeture hermétique et éliminés conformément à la réglementation en vigueur. Les mains doivent ensuite être soigneusement lavées au savon et à l’eau. Les femmes enceintes ou qui pensent l’être ne doivent pas manipuler la plaquette ou la gélule.
· Les gélules peuvent être prises au cours ou en dehors des repas.
· Vous devez prendre LENALIDOMIDE STRAGEN 10 mg, gélule à peu près à heure fixe les jours prévus de chaque cycle.
Prise de ce médicament
Pour sortir la gélule de la plaquette :
· Appuyez seulement sur une extrémité de la gélule pour la pousser à travers la pellicule d’aluminium.
· N’exercez pas de pression sur le centre de la gélule car cela peut provoquer sa rupture.
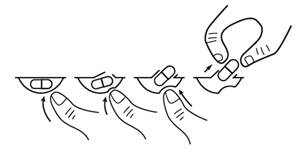
Durée du traitement par LENALIDOMIDE STRAGEN 10 mg, gélule
LENALIDOMIDE STRAGEN 10 mg, gélule est pris en suivant des cycles de traitement d’une durée de 21 ou 28 jours chacun (voir la section « Cycle de traitement » ci-dessus). Vous devez poursuivre les cycles de traitement jusqu’à ce que votre médecin vous dise d’arrêter.
Si vous avez pris plus de LENALIDOMIDE STRAGEN10 mg, gélule que vous n’auriez dû
Si vous avez pris plus de LENALIDOMIDE STRAGEN 10 mg, gélule que ce qui vous a été prescrit, prévenez immédiatement votre médecin.
Si vous oubliez de prendre LENALIDOMIDE STRAGEN 10 mg, gélule
Si vous avez oublié de prendre LENALIDOMIDE STRAGEN 10 mg, gélule à l’heure habituelle et :
· Si moins de 12 heures se sont écoulées - prenez la gélule immédiatement.
· Si plus de 12 heures se sont écoulées - ne prenez pas la gélule. Prenez la gélule suivante à l’heure habituelle, le lendemain.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin ou à votre pharmacien.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
· Urticaire, éruptions cutanées, gonflement des yeux, de la bouche ou du visage, difficultés à respirer ou démangeaisons, qui peuvent être des symptômes de formes graves de réaction allergiques appelées angio-œdème et réaction anaphylactique
· Réaction allergique grave qui peut débuter sous forme d’éruption cutanée sur une seule zone, mais qui progresse avec un décollement de la peau s’étendant à tout le corps (syndrome de Stevens-Johnson et/ou nécrolyse épidermique toxique)
· Eruption cutanée étendue, fièvre élevée, augmentations des enzymes hépatiques, anomalies du sang (éosinophilie), augmentation du volume des ganglions lymphatiques et atteinte d’autres organes (réaction médicamenteuse accompagnée d’une éosinophilie et de symptômes systémiques, appelée également syndrome DRESS ou syndrome d’hypersensibilité médicamenteuse). Voir également la rubrique 2.
Informez immédiatement votre médecin si vous remarquez l’un des effets indésirables graves suivants :
· Fièvre, frissons, mal de gorge, toux, aphtes ou autres symptômes d’infection, y compris dans le sang (sepsis)
· Saignement ou ecchymose (bleu) en l’absence de blessure
· Douleur dans la poitrine ou la jambe
· Essoufflement
· Douleurs osseuses, faiblesse musculaire, confusion ou fatigue pouvant être la conséquence d’un taux élevé de calcium dans le sang
LENALIDOMIDE STRAGEN 10 mg, gélule peut diminuer le nombre des globules blancs qui sont des cellules luttant contre les infections, ainsi que celui des cellules sanguines qui contribuent à faire coaguler le sang (les plaquettes), ce qui peut entraîner des problèmes de saignements, comme des saignements de nez et des ecchymoses (« bleus »).
LENALIDOMIDE STRAGEN 10 mg, gélule peut également entraîner la formation de caillots sanguins dans les veines (thrombose).
Autres effets indésirables
Il est important de noter qu’un faible nombre de patients peut développer d’autres types de cancers et il est possible que ce risque soit majoré en cas de traitement par LENALIDOMIDE STRAGEN 10 mg, gélule. Par conséquent, votre médecin devra évaluer attentivement le bénéfice et le risque quand il vous prescrit LENALIDOMIDE STRAGEN 10 mg, gélule.
Effets indésirables très fréquents (pouvant affecter plus de 1 patient sur 10)
· Chute du nombre de globules rouges, ce qui peut provoquer une anémie entraînant fatigue et faiblesse
· Eruptions cutanées, démangeaisons
· Crampes musculaires, faiblesse musculaire, douleurs musculaires, douleurs osseuses, douleurs articulaires, douleurs dorsales, douleurs dans les extrémités
· Gonflement généralisé, y compris gonflement des bras et des jambes
· Faiblesse, fatigue
· Fièvre et symptômes grippaux, notamment fièvre, douleur musculaire, maux de tête, douleur dans l’oreille, toux et frissons
· Engourdissement, fourmillements ou sensation de brûlure de la peau, douleurs aux mains ou aux pieds, vertiges, tremblements
· Diminution de l’appétit, modifications du goût
· Augmentation de la douleur, de la taille de la tumeur, ou rougeur autour de la tumeur
· Perte de poids
· Constipation, diarrhée, nausées, vomissements, douleur gastrique, brûlures d’estomac
· Faible taux de potassium ou de calcium et/ou de sodium dans le sang
· Fonction thyroïdienne inférieure à ce qu’elle devrait être
· Douleur dans la jambe (pourrait être un signe de thrombose), douleur dans la poitrine ou essoufflement (peut être un signe de la présence de caillots de sang dans les poumons, appelée embolie pulmonaire)
· Infections de tous types y compris infection des sinus qui entourent le nez, infection des poumons et des voies respiratoires supérieures
· Essoufflement
· Vision trouble
· Opacification de l’œil (cataracte)
· Problèmes rénaux incluant une fonction rénale diminuée ou une incapacité des reins à maintenir une fonction normale
· Anomalies des analyses biologiques du foie
· Augmentation des taux d’enzymes hépatiques
· Modification d’une protéine présente dans le sang qui peut provoquer un gonflement des artères (vascularite)
· Augmentation du taux de sucre dans le sang (diabète)
· Diminution du taux de sucre dans le sang
· Maux de tête
· Saignements de nez
· Peau sèche
· Dépression, modifications de l’humeur, troubles du sommeil
· Toux
· Baisse de la pression artérielle
· Vague sensation de gêne corporelle, sensation de mal-être
· Ulcérations dans la bouche, bouche sèche
· Déshydratation
Effets indésirables fréquents (pouvant affecter jusqu’à 1 patient sur 10)
· Destruction des globules rouges (anémie hémolytique)
· Certains types de cancers de la peau
· Saignement des gencives, de l’estomac ou des intestins
· Augmentation de la pression artérielle, battements de cœur lents, rapides ou irréguliers
· Augmentation d’une substance provenant de la destruction normale ou anormale des globules rouges
· Augmentation d’un type de protéine qui est un signe d’inflammation dans l’organisme
· Coloration plus foncée de la peau, coloration anormale de la peau résultant des saignements se produisant sous la peau, généralement dus à des ecchymoses (bleus), formation de vésicules remplies de sang sur la peau, ecchymoses (bleus)
· Augmentation du taux d’acide urique dans le sang
· Eruptions cutanées, rougeur de la peau, fissures sur la peau, présence de squames sur la peau ou peau qui pèle, urticaire
· Augmentation de la transpiration, sueurs nocturnes
· Difficulté à avaler, mal de gorge, altération de la qualité de la voix ou changements de voix
· Nez qui coule
· Production d’urine plus importante ou plus faible qu’habituellement ou difficulté à contrôler les mictions
· Présence de sang dans les urines
· Essoufflement, en particulier au moment de s’allonger (pouvant être le symptôme d’une insuffisance cardiaque)
· Difficulté à avoir une érection
· Accident vasculaire cérébral, évanouissement, vertige (problème au niveau de l’oreille interne, avec pour conséquence la sensation que tout tourne autour de soi), perte de connaissance temporaire
· Douleur dans la poitrine irradiant vers les bras, le cou, la mâchoire, le dos ou l’estomac, sueur et essoufflement, nausées ou vomissements, pouvant être les signes d’une crise cardiaque (infarctus du myocarde)
· Faiblesse musculaire, manque d’énergie
· Douleur dans le cou, douleur dans la poitrine
· Frissons
· Gonflement des articulations
· Ralentissement ou blocage de l’écoulement de la bile à partir du foie
· Baisse des taux de phosphate ou de magnésium dans le sang
· Difficultés pour parler
· Atteinte hépatique
· Troubles de l’équilibre, difficulté à se mouvoir
· Surdité, bourdonnements dans les oreilles (acouphènes)
· Douleurs d’origine neurologique, sensations anormales désagréables, en particulier au toucher Surcharge en fer
· Soif
· Confusion
· Mal de dents
· Chute pouvant être à l’origine de fractures
Effets indésirables peu fréquents (pouvant affecter jusqu’à 1 patient sur 100)
· Saignements dans le crâne
· Problèmes de circulation
· Perte de vision
· Perte de désir sexuel (libido)
· Urine émise en quantité importante avec douleur osseuse et faiblesse, pouvant être des signes d‘une maladie des reins (syndrome de Fanconi)
· Coloration jaune de la peau, des membranes ou des yeux (ictère ou « jaunisse »), selles claires, urines sombres, démangeaisons de la peau, éruption cutanée, douleur au niveau de l’estomac ou ballonnements, qui peuvent être des symptômes d’atteinte du foie (affection hépatique)
· Douleur abdominale, ballonnement ou diarrhée, pouvant être des signes d’inflammation du gros intestin (appelée colite ou caecite)
· Atteinte des cellules rénales (appelée nécrose tubulaire rénale)
· Modifications de la couleur de la peau, sensibilité à la lumière du soleil
· Syndrome de lyse tumorale – des complications métaboliques peuvent survenir pendant le traitement du cancer et même parfois sans traitement. Ces complications sont causées par les produits de dégradation des cellules cancéreuses qui sont détruites. Elles peuvent inclure : des modifications du bilan sanguin ; taux élevés de potassium, de phosphore, d’acide urique et faibles taux de calcium entraînant des modifications de la fonction rénale et du rythme cardiaque, des convulsions et dans certains cas le décès
Effets indésirables de fréquence indéterminée (ne peut être estimée à partir des données disponibles)
· Douleur soudaine ou douleur légère allant en s’intensifiant, dans le haut de l’estomac et (ou) dans le dos, persistant plusieurs jours et pouvant s’accompagner de nausées, vomissements, fièvre et d’un pouls rapide. Ces symptômes peuvent être dus à l’inflammation du pancréas.
· Respiration sifflante, essoufflement ou toux sèche, pouvant être le signe de l’inflammation du tissu pulmonaire
· De rares cas de destruction du tissu musculaire (douleur, faiblesse ou gonflement musculaires) pouvant entraîner des troubles rénaux (« rhabdomyolyse ») ont été observés, dont certains lorsque LENALIDOMIDE STRAGEN 10 mg, gélule est administré avec une statine (un type de médicament utilisé pour faire diminuer le taux de cholestérol)
· Affection de la peau causée par l’inflammation de petits vaisseaux sanguins, accompagnée de douleurs articulaires et de fièvre (vascularite leucocytoclasique)
· Perforation de la paroi de l’estomac ou de l'intestin. Cela peut entraîner une infection très grave. Informez votre médecin si vous présentez des douleurs d’estomac intenses, de la fièvre, des nausées, des vomissements, du sang dans les selles ou des modifications des selles
· Infections virales, y compris zona (une maladie virale qui provoque une éruption cutanée douloureuse avec des vésicules) et récidive de l’hépatite B (qui peut provoquer un jaunissement de la peau et du blanc de l’œil, des urines foncées, des douleurs dans le côté droit de l’abdomen, une fièvre et des nausées ou vomissements)
· Rejet du greffon après une transplantation d’organe (tel qu’un rein, le cœur)
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou à votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER LENALIDOMIDE STRAGEN 10 mg, gélule ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur la plaquette et l’emballage après « EXP ». La date de péremption fait référence au dernier jour de ce mois.
Ce médicament ne nécessite pas de précautions particulières de conservation.
N’utilisez pas ce médicament si vous remarquez que l’emballage est endommagé ou a été ouvert.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Rapportez les gélules non utilisées à votre pharmacien. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient LENALIDOMIDE STRAGEN 10 mg, gélule
· La substance active est : lénalidomide.
Chaque gélule contient 10 mg de lénalidomide.
· Les autres composants sont :
Contenu de la gélule : lactose, cellulose microcristalline (E460(i)), croscarmellose sodique (E468) et stéarate de magnésium (E470b)
Enveloppe de la gélule : gélatine, dioxyde de titane (E171), indigotine (E132) et oxyde de fer jaune (E172)
Encre d’impression : gomme laque (E904), propylène glycol (E1520), hydroxyde de potassium (E525) et oxyde de fer noir (E172)
Qu’est-ce que LENALIDOMIDE STRAGEN 10 mg, gélule, et contenu de l’emballage extérieur
LENALIDOMIDE STRAGEN 10 mg, gélule avec un corps jaune opaque et une tête verte à vert clair opaque, avec une longueur d’approximativement 21,7 mm, portant l’inscription « L9NL » et « 10 ».
Boîte de 7, 14, 21, 28 et 42 gélules sous plaquettes (OPA/Aluminium/PVC/Aluminium).
Titulaire de l’autorisation de mise sur le marché
30 RUE EDOUARD NIEUPORT
69008 LYON
Exploitant de l’autorisation de mise sur le marché
STRAGEN FRANCE
30 RUE EDOUARD NIEUPORT
69008 LYON
C/ CASTELLO N°1,
POL. LAS SALINAS,
08830 SANT BOI DE LLOBREGAT (BARCELONE)
ESPAGNE
OU
SYNTHON B.V.
MICROWEG 22
6545 CM, NIJMEGEN
PAYS-BAS
OU
CENTRE LAB
ZONE ARTISANALE GRANDERAIE
23000 GUERET
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Ce médicament est autorisé dans les Etats membres de l'Espace Economique Européen sous les noms suivants : Conformément à la réglementation en vigueur.
[À compléter ultérieurement par le titulaire]
La dernière date à laquelle cette notice a été révisée est :
[À compléter ultérieurement par le titulaire]
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).