FEBUXOSTAT ARROW
Dosage: 80mg, 120mg.

")
")
. La carte Vitale est une marque déposée par l'assurance maladie")
Cliquez sur un pictogramme pour aller directement à la rubrique le concernant.
Pour plus d'information sur les pictogrammes, consultez l'aide.
Informations importantes 
Les informations importantes disponibles pour ce médicament sont les suivantes :
Indications thérapeutiques
FÉBUXOSTAT ARROW agit en réduisant le taux d’acide urique dans le sang (uricémie). Le maintien de l’uricémie à un niveau bas, grâce à la prise quotidienne de FÉBUXOSTAT ARROW, permet d’arrêter l'accumulation de cristaux et de réduire progressivement les symptômes. Le maintien d’une uricémie suffisamment basse pendant une durée suffisamment longue peut également permettre une diminution de la taille des tophi.
FEBUXOSTAT ARROW comprimé à 120 mg est aussi utilisé pour traiter et éviter une élévation du taux sanguin d’acide urique, qui peut apparaître quand on commence à recevoir une chimiothérapie pour traiter les cancers du sang.
Quand on reçoit une chimiothérapie, les cellules cancéreuses sont détruites, et par conséquent le taux d’acide urique augmente dans le sang, à moins que l’on empêche la formation d’acide urique.
FÉBUXOSTAT ARROW est indiqué chez l’adulte.
Groupe(s) générique(s)
Ce médicament appartient au(x) groupe(s) générique(s) suivants :
Composition en substances actives
-
 Comprimé (Composition pour un comprimé)
Comprimé (Composition pour un comprimé)
-
> fébuxostat
 80 mg
80 mg
-
> fébuxostat
Présentations
> plaquettes transparentes PVC aluminium de 28 comprimés
Code CIP : 34009 301 709 0 8Déclaration de commercialisation : 25/04/2019
Cette présentation est agréée aux collectivités
Taux de remboursement : 65%
Service médical rendu (SMR) 
Ce médicament étant un générique, le SMR n'a pas été évalué par la commission de la transparence (CT), il est possible de se référer à la /aux spécialité(s) de référence du groupe générique auquel appartient ce médicament (cliquez ici pour aller à la rubrique des groupes génériques)
Amélioration du service médical rendu (ASMR)
Ce médicament étant un générique, l'ASMR n'a pas été évalué par la commission de la transparence (CT), il est possible de se référer à la /aux spécialité(s) de référence du groupe générique auquel appartient ce médicament (cliquez ici pour aller à la rubrique des groupes génériques)
Autres informations (cliquer pour afficher)
- Titulaire de l'autorisation : ARROW GENERIQUES
- Conditions de prescription et de délivrance :
- Statut de l'autorisation : Valide
- Type de procédure : Procédure décentralisée
- Code CIS : 6 150 990 9
ANSM - Mis à jour le : 02/05/2022
FÉBUXOSTAT ARROW 80 mg, comprimé pelliculé
2. COMPOSITION QUALITATIVE ET QUANTITATIVE 
Fébuxostat (sous forme hémihydratée) ................................................................................... 80 mg
Pour un comprimé pelliculé.
Excipient à effet notoire : chaque comprimé contient 76,5 mg de lactose (sous forme monohydratée).
Pour la liste complète des excipients, voir rubrique 6.1.
Comprimé pelliculé biconvexe de forme ovale, jaune pâle à jaune, portant l’inscription « FEB » sur une face et « 80 » sur l’autre face. La taille des comprimés est de 14,7mm x 8,7mm.
4.1. Indications thérapeutiques
Le fébuxostat est indiqué chez l’adulte.
4.2. Posologie et mode d'administration
La dose recommandée de fébuxostat est de 80 mg une fois par jour, administrée par voie orale, pendant ou en dehors des repas. Si l’uricémie est > 6 mg/dL (357 µmol/L) après deux à quatre semaines de traitement, l‘administration de fébuxostat 120 mg une fois par jour peut être envisagée.
L’action de fébuxostat est suffisamment rapide pour permettre un nouveau dosage de l’uricémie après deux semaines de traitement. L’objectif thérapeutique est la diminution et le maintien de l’uricémie au-dessous de 6 mg/dL (357μmol/L).
Un traitement préventif des crises de goutte est recommandé pendant au moins six mois (voir rubrique 4.4).
Sujet âgé
Aucune adaptation posologique n’est nécessaire chez les patients âgés (voir rubrique 5.2).
Insuffisance rénale
L’efficacité et la tolérance n’ont pas été totalement évaluées chez les patients présentant une insuffisance rénale sévère (clairance de la créatinine < 30 mL/min) (voir rubrique 5.2).
Aucune adaptation posologique n’est nécessaire chez les patients présentant une insuffisance rénale légère à modérée.
Insuffisance hépatique
L’efficacité et la tolérance du fébuxostat n’ont pas été étudiées chez les patients présentant une insuffisance hépatique sévère (classe C de Child Pugh).
La dose recommandée est de 80 mg chez les patients présentant une insuffisance hépatique légère. L’expérience clinique est limitée chez les patients présentant une insuffisance hépatique modérée.
Population pédiatrique
La sécurité et l’efficacité du fébuxostat chez les enfants de moins de 18 ans n’ont pas été établies. Aucune donnée n’est disponible.
Mode d’administration
Voie orale.
FÉBUXOSTAT ARROW doit être pris par voie orale et peut être pris au cours ou en dehors des repas.
4.4. Mises en garde spéciales et précautions d'emploi
Chez les patients déjà atteints de maladies cardiovasculaires sévères (par exemple : infarctus du myocarde, AVC ou angor instable) pendant le développement du produit et dans une étude post-AMM (CARES), un nombre plus élevé d’évènements cardiovasculaires fatals a été observé avec le fébuxostat comparativement à l’allopurinol.
Cependant, dans une étude post-AMM (FAST) plus récente, le fébuxostat a été inférieur à l’allopurinol sur l’incidence des évènements cardiovasculaires fatals et non fatals. Le traitement de ce groupe de patients doit être pratiqué avec prudence et ces patients doivent être surveillés régulièrement. Pour plus de détails sur la tolérance cardiovasculaire du fébuxostat, se reporter aux sections 4.8 et 5.1.
Allergie au médicament/hypersensibilité
De rares cas de graves réactions allergiques/d’hypersensibilité, incluant des syndromes de Stevens- Johnson pouvant être fatal, des nécrolyses épidermiques toxiques (Syndrome de Lyell) et des réactions/chocs anaphylactiques aigus, ont été rapportés après commercialisation. Dans la plupart des cas, ces réactions sont survenues durant le premier mois de traitement par fébuxostat. Pour certains de ces patients, il a été rapporté une insuffisance rénale et/ou un antécédent d’hypersensibilité à l’allopurinol. Dans certains cas, les réactions graves d’hypersensibilité dont le syndrome d’hypersensibilité médicamenteuse avec éosinophilie et symptômes systémiques (syndrome DRESS), étaient associées à de la fièvre, une atteinte hématologique, rénale ou hépatique.
Les patients doivent être informés des signes et symptômes des réactions allergiques/d’hypersensibilité et doivent être étroitement surveillés au regard de ces symptômes (voir rubrique 4.8). Le traitement par fébuxostat doit être immédiatement arrêté en cas de survenue de réactions graves allergiques/d’hypersensibilité, incluant le syndrome de Stevens-Johnson, étant donné que l’arrêt précoce du traitement est associé à un meilleur pronostic. Si le patient a développé une réaction allergique/d’hypersensibilité incluant le syndrome de Stevens-Johnson ou une réaction/choc anaphylactique aigu, le traitement par fébuxostat ne doit jamais être réinstauré.
Crise de goutte
Le traitement par fébuxostat ne doit pas être instauré avant la disparition complète d’une crise de goutte. Des crises de goutte peuvent survenir en début de traitement en raison d’une variation de l’uricémie qui entraîne une mobilisation des cristaux d’urate à partir des dépôts tissulaires (voir rubriques 4.8 et 5.1). Lors de l’instauration d’un traitement par fébuxostat, un traitement préventif de la crise de goutte par un anti-inflammatoire non stéroïdien ou par la colchicine est recommandé pendant au moins six mois (voir rubrique 4.2).
En cas de survenue d'une crise de goutte au cours du traitement, ne pas interrompre la prise de fébuxostat. Un traitement de la crise de goutte adapté à chaque patient doit être administré simultanément. La fréquence et l’intensité des crises de goutte diminuent lors de la poursuite du traitement par fébuxostat.
Dépôt de xanthine
Chez les patients ayant une production d’urate fortement accrue (par exemple affection maligne traitée, syndrome de Lesch-Nyhan), la concentration absolue de xanthine au niveau urinaire peut, dans de rares cas, augmenter suffisamment pour entraîner un dépôt dans les voies urinaires. En l’absence d’expérience clinique avec le fébuxostat dans cette population, son administration n’est pas recommandée chez ces patients.
Mercaptopurine/azathioprine
L’administration du fébuxostat n’est pas recommandée chez les patients traités par mercaptopurine/azathioprine car l’inhibition de la xanthine oxydase par le fébuxostat peut entraîner une augmentation des concentrations plasmatiques de mercaptopurine/azathioprine qui peut provoquer une toxicité sévère.
Si cette association ne peut être évitée, une diminution de la posologie de mercaptopurine/azathioprine à 20 % ou moins de la dose préalablement prescrite est recommandée afin d’éviter les possibles effets hématologiques (voir les rubriques 4.5 et 5.3).
Les patients doivent être étroitement surveillés et la dose de mercaptopurine/azathioprine doit être ajustée en conséquence sur la base de l'évaluation de la réponse thérapeutique et de l'apparition d'éventuels effets toxiques.
Greffe d’organe
En l’absence d’expérience clinique chez le patient ayant reçu une greffe d’organe, l’utilisation de fébuxostat n’est pas recommandée chez ces patients (voir rubrique 5.1).
Théophylline
L’administration concomitante de fébuxostat 80 mg et de théophylline 400 mg en dose unique à des sujets sains a démontré l’absence de toute interaction pharmacocinétique (voir rubrique 4.5). Le fébuxostat 80 mg peut être prescrit chez les patients traités par théophylline sans risque d’augmentation des concentrations plasmatiques de théophylline. Aucune donnée n’est disponible pour le fébuxostat 120 mg.
Affections hépatiques
Les résultats combinés des études cliniques de phase III ont montré de légères anomalies du bilan hépatique chez des patients (5,0 %) traités par fébuxostat. La réalisation d’un bilan hépatique est recommandée avant l’instauration du traitement par fébuxostat et périodiquement par la suite, en fonction du jugement clinique (voir rubrique 5.1).
Affections de la thyroïde
Au cours des études d’extension en ouvert à long terme, une augmentation du taux de TSH (> 5,5 µUI/mL) a été observée chez des patients traités au long cours par fébuxostat (5,5 %). Le fébuxostat doit être prescrit avec prudence chez les patients présentant une altération de la fonction thyroïdienne (voir rubrique 5.1).
Lactose
Les comprimés de fébuxostat contiennent du lactose. Les patients présentant des troubles héréditaires rares d’intolérance au galactose, un déficit total en lactase ou un syndrome de malabsorption du glucose et du galactose ne doivent pas prendre ce médicament.
Sodium
Ce médicament contient moins de 1 mmol (23 mg) de sodium par comprimé pelliculé, c’est-à-dire qu’il est essentiellement « sans sodium ».
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Mercaptopurine/azathioprine
En raison de son mécanisme d’action inhibiteur de la Xanthine Oxydase (XO), l’administration concomitante de fébuxostat n’est pas recommandée. L’inhibition de la XO par le fébuxostat peut entraîner une augmentation des concentrations plasmatiques de ces médicaments et provoquer une myélotoxicité.
En cas de co-administration avec le fébuxostat, la dose de mercaptopurine/azathioprine doit être réduite à 20 % ou moins de la dose préalablement prescrite (voir les rubriques 4.4 et 5.3).
L’adéquation et l’ajustement posologique proposé, qui reposait sur une analyse de modélisation et de simulation à partir de données précliniques chez le rat, a été confirmée par les résultats d’une étude clinique d’interaction médicamenteuse chez des volontaires sains, recevant 100 mg d’azathioprine seule et une dose réduite d’azathioprine (25 mg) en association avec le fébuxostat (40 ou 120 mg).
Aucune étude d’interaction médicamenteuse du fébuxostat avec d’autres chimiothérapies cytotoxiques n’a été menée. Aucune donnée n’est disponible quant à la sécurité d’emploi du fébuxostat en association avec d’autres traitements cytotoxiques.
Rosiglitazone/substrats du CYP2C8
Il a été montré que le fébuxostat était un inhibiteur faible du CYP2C8 in vitro. Dans une étude chez des sujets sains, l’administration concomitante de 120 mg de fébuxostat une fois par jour et de 4 mg de rosiglitazone en prise unique par voie orale, n’a eu aucun effet sur la pharmacocinétique de la rosiglitazone ni sur son métabolite, le N-desmethyl rosiglitazone, indiquant que le fébuxostat n’est pas un inhibiteur de l’enzyme CYP2C8 in vivo. Ainsi, l’administration concomitante de fébuxostat et de rosiglitazone ou d’autres substrats du CYP2C8 ne devrait pas nécessiter d’ajustement de la posologie de ces produits.
Théophylline
Une étude d’interaction chez des sujets sains a été menée avec le fébuxostat afin d’évaluer si l’inhibition de la XO peut induire une élévation des concentrations de théophylline circulante, comme cela a été décrit avec d'autres inhibiteurs de la XO. Les résultats de l’étude ont montré que l’administration concomitante de fébuxostat 80 mg une fois par jour et de théophylline 400 mg en dose unique n’a pas d’effet sur la pharmacocinétique et la sécurité de la théophylline. Aucune précaution particulière n’est donc recommandée en cas d’administration concomitante de fébuxostat 80 mg et de théophylline. Aucune donnée n’est disponible concernant le fébuxostat 120 mg.
Naproxène et autres inhibiteurs de la glycuronidation
Le métabolisme du fébuxostat dépend des enzymes Uridine Glucuronyl Transférase (UGT). Les médicaments qui inhibent la glycuronidation, tels les anti-inflammatoires non stéroïdiens et le probénécide, pourraient théoriquement affecter l’élimination du fébuxostat. Chez des volontaires sains, l’administration concomitante de fébuxostat et de naproxène 250 mg deux fois par jour a été associée à une augmentation de l’exposition au fébuxostat (Cmax 28 %, ASC 41 % et t1/2 26 %). Au cours des études cliniques, l’administration de naproxène ou d’autres anti-inflammatoires non stéroïdiens ou inhibiteurs de la Cox 2 n'a pas été associée à une augmentation cliniquement significative des événements indésirables.
Le fébuxostat peut être administré de façon concomitante avec le naproxène sans qu’une adaptation de la posologie du fébuxostat ou du naproxène ne soit nécessaire.
Inducteurs de la glycuronidation
Les inducteurs puissants des enzymes UGT peuvent accroître le métabolisme et diminuer l’efficacité du fébuxostat. Un contrôle de l’uricémie est donc recommandé une à deux semaines après le début d’un traitement par un inducteur puissant de la glycuronidation. A l’inverse, l’arrêt du traitement par un inducteur pourrait se traduire par une augmentation de la concentration plasmatique du fébuxostat.
Colchicine/indométacine/hydrochlorothiazide/warfarine
Le fébuxostat peut être administré de façon concomitante avec la colchicine ou l’indométacine sans adaptation de la dose de l’une ou l’autre des substances actives.
Aucune adaptation posologique du fébuxostat n’est nécessaire en cas d’administration concomitante d’hydrochlorothiazide.
Aucune adaptation posologique de la warfarine n’est nécessaire en cas d’administration concomitante avec le fébuxostat. L’administration concomitante de fébuxostat (80 mg ou 120 mg en une prise par jour) et de warfarine n’a pas montré d’effet sur la pharmacocinétique de la warfarine chez des sujets sains. L’INR et l’activité du facteur VII n’ont pas non plus été affectés par la co-administration de fébuxostat.
Désipramine/substrats du CYP2D6
Le fébuxostat exerce un léger effet inhibiteur du CYP2D6 in vitro. Lors d’une étude chez le volontaire sain, l’administration de 120 mg de fébuxostat une fois par jour a conduit à une augmentation moyenne de 22 % de l’ASC de la désipramine, substrat du CYP2D6, témoignant d’un faible effet inhibiteur potentiel du fébuxostat sur le CYP2D6 in vivo. L’administration concomitante de fébuxostat avec d’autres substrats du CYP2D6 ne devrait donc pas nécessiter d’adaptation de la posologie de ces produits.
Antiacides
La prise concomitante d’un antiacide contenant des hydroxydes de magnésium et d’aluminium a retardé l’absorption du fébuxostat (d’environ une heure) et a induit une diminution de 32 % de la Cmax, mais sans modification significative de l’ASC. Le fébuxostat peut donc être administré sans tenir compte de la prise concomitante d’un anti-acide.
4.6. Fertilité, grossesse et allaitement
Les données recueillies sur un nombre très limité de grossesses n'ont pas révélé d’effet délétère du fébuxostat sur la grossesse ou sur le fœtus/nouveau-né. Les études menées chez l'animal n’ont pas montré d'effets délétères directs ou indirects sur la gestation, le développement embryonnaire ou fœtal ou la mise bas (voir rubrique 5.3). Le risque potentiel en clinique n'est pas connu. Le fébuxostat ne doit pas être utilisé au cours de la grossesse.
Allaitement
L’excrétion du fébuxostat dans le lait maternel n’est pas connue. Des études menées chez l’animal ont montré une excrétion du principe actif dans le lait et une altération du développement des petits allaités. Un risque pour le nourrisson allaité ne peut être exclu. Le fébuxostat ne doit pas être utilisé chez la femme qui allaite.
Fertilité
Les études de reproduction chez l’animal à des doses allant jusqu’à 48 mg/kg/jour n’ont pas montré d’effets néfastes dose dépendant sur la fécondité (voir rubrique 5.3). L’effet du fébuxostat sur la fécondité chez l’homme n’est pas connu.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Résumé du profil de sécurité
Les effets indésirables les plus fréquemment rapportés au cours des études cliniques (4 072 patients traités par au moins une dose de 10 mg à 300 mg), des études de sécurité post-AMM (étude FAST : 3001 patients traités avec une dose de 80 mg à 120 mg) et après commercialisation sont des crises de gouttes, des anomalies de la fonction hépatique, des diarrhées, des nausées, des maux de tête, des sensations vertigineuses, des dyspnées, des éruptions, des prurits, des arthralgies, des myalgies, des douleurs aux extrémités, des œdèmes et de la fatigue. Ces effets indésirables étaient généralement de sévérité légère ou modérée. De rares réactions graves d’hypersensibilité au fébuxostat, dont certaines étaient associées à des symptômes généraux, ainsi que des évènements rares de mort subite cardiaque, ont été observées après commercialisation.
Liste tabulée des effets indésirables
Les effets indésirables fréquents (≥ 1/100 à < 1/10), peu fréquents (≥ 1/1 000 à < 1/100) et rares (≥ 1/10 000 à < 1/1 000), survenant chez les patients traités par fébuxostat sont mentionnés ci-dessous.
Dans chaque groupe de fréquence, les effets indésirables sont présentés par ordre de sévérité décroissante.
Tableau 1 : effets indésirables lors des études de phase III, des études d’extension à long terme, des études de sécurité post-AMM et après commercialisation
|
Affections hématologiques et du système lymphatique |
Rare Pancytopénie, thrombocytopénie, agranulocytose*, anémie# |
|
Affections du système immunitaire |
Rare Réaction anaphylactique*, hypersensibilité médicamenteuse* |
|
Troubles endocriniens |
Peu fréquent TSH sanguine augmentée, hypothyroïdie# |
|
Affections oculaires |
Peu fréquent Vision trouble Rare Occlusion de l’artère rétinienne# |
|
Troubles du métabolisme et de la nutrition |
Fréquent*** Crises de goutte Peu fréquent Diabète sucré, hyperlipidémie, diminution de l’appétit, prise de poids Rare Perte de poids, augmentation de l’appétit, anorexie |
|
Affections psychiatriques |
Peu fréquent Diminution de la libido, insomnie Rare Nervosité, humeur dépressive#, troubles du sommeil# |
|
Affections du système nerveux |
Fréquent Maux de tête, sensations vertigineuses Peu fréquent Paresthésies, hémiparésie, somnolence, léthargie#, altération du goût, hypoesthésie, hyposmie Rare Agueusie#, sensation de brûlure# |
|
Affections auditives et du labyrinthe |
Peu fréquent Acouphènes Rare Vertige# |
|
Affections cardiaques |
Peu fréquent Fibrillation auriculaire, palpitations, anomalies de l’ECG, arythmies# Rare Mort subite cardiaque* |
|
Affections vasculaires |
Peu fréquent Hypertension, bouffées vasomotrices, bouffées de chaleur Rare Collapsus circulatoire# |
|
Affections respiratoires |
Fréquent Dyspnée Peu fréquent Bronchite, infections des voies respiratoires supérieures, infections des voies respiratoires inférieures#, toux, rhinorrhées# Rare Pneumonie# |
|
Affections gastro-intestinales |
Fréquent Diarrhées**, nausées Peu fréquent Douleurs abdominales, douleurs abdominales hautes#, distension abdominale, reflux gastro- œsophagien, vomissements, sécheresse buccale, dyspepsie, constipation, selles fréquentes, flatulences, gêne gastro-intestinale, ulcération buccale, gonflement des lèvres#, pancréatite Rare Performation gastro-intestinale#, stomatite# |
|
Affections hépatobiliaires |
Fréquent Anomalies du bilan hépatique** Peu fréquent Cholélithiase Rare Hépatite, jaunisse*, lésion du foie*, cholécystite# |
|
Affections de la peau et du tissu sous-cutané |
Fréquent Éruptions (incluant éruptions de type varié rapportées avec une fréquence plus faible, voir ci-dessous), prurit Peu fréquent Dermatite, urticaire, décoloration de la peau, lésions cutanées, pétéchie, éruption maculaire, éruption maculo-papuleuse, éruption papuleuse, hyperhidrose, alopécie, eczéma#, érythème, sueurs nocturnes#, psoriasis#, rash prurigineux# Rare Nécrolyse épidermique toxique (syndrome de Lyell)*, syndrome de Stevens-Johnson*, angiœdème*, syndrome d’hypersensibilité médicamenteuse avec éosinophilie et symptômes systémiques (syndrome DRESS)*, éruption généralisée (grave)*, éruption exfoliative, éruption folliculaire, éruption vésiculaire, éruption pustuleuse, éruption érythémateuse, éruption morbilliforme |
|
Affections musculo-squelettiques et systémiques |
Fréquent Arthralgie, myalgies, extrémités douloureuses# Peu fréquent Arthrite, douleurs musculo-squelettiques, faiblesse musculaire, spasmes musculaires, contracture musculaire, bursite, tuméfaction articulaire#, dorsalgie#, raideur musculo-squelettique#, raideur articulaire Rare Rhabdomyolyse*, syndrome de la coiffe des rotateurs#, pseudopolyarthrite rhizomélique# |
|
Affections du rein et des voies urinaires |
Peu fréquent Insuffisance rénale, lithiase rénale, hématurie, pollakiurie, protéinurie, miction impérieuse, infection des voies urinaires# Rare Néphrite tubulo-interstitielle* |
|
Affections du système de reproduction et des seins |
Peu fréquent Dysfonction érectile |
|
Troubles généraux et anomalies au site d’administration |
Fréquent Œdème, fatigue Peu fréquent Douleurs thoraciques, gêne dans la poitrine, douleur#, malaise# Rare Soif, sensation de chaud# |
|
Modifications des paramètres biologiques |
Peu fréquent Augmentation de l’amylasémie, diminution de la numération plaquettaire, diminution du nombre de globules blancs, diminution du nombre de lymphocytes, augmentation de la créatininémie, diminution de l’hémoglobinémie, augmentation de l’urémie, augmentation de la triglycéridémie, augmentation de la cholestérolémie, diminution de l’hématocrite, augmentation de la lactate déshydrogénase dans le sang, augmentation de la kaliémie, élévation de l’INR# Rare Augmentation de la glycémie, allongement du temps de céphaline activée, diminution des globules rouges, augmentation des phosphatases alcalines dans le sang, augmentation des créatine phosphokinases dans le sang* |
|
Lésions, intoxications, et complications d’interventions |
Peu fréquent Contusion# |
* effets indésirables liés au traitement issus des données après commercialisation
** Les résultats combinés des études de phase 3 ont montré des diarrhées non infectieuses et des anomalies de la fonction hépatique plus fréquentes chez les patients traités de façon concomitante par la colchicine.
*** Voir rubrique 5.1 pour l’incidence des crises de goutte dans les études de phase III randomisées et contrôlées.
# Effets indésirables provenant des études de sécurité post-AMM
Description des événements indésirables spécifiques
De rares réactions graves d’hypersensibilité au fébuxostat, incluant le syndrome de Stevens-Johnson, une nécrolyse épidermique toxique (syndrome de Lyell), et de réaction/choc anaphylactique ont été observées après commercialisation. Le syndrome de Stevens-Johnson et la nécrolyse épidermique toxique sont caractérisés par une éruption cutanée progressive, accompagnée de bulles ou de lésions des muqueuses et une irritation oculaire. Les réactions d’hypersensibilité au fébuxostat peuvent être associées aux symptômes suivants : réactions cutanées caractérisées par une éruption maculo- papuleuse infiltrée, une éruption généralisée ou exfoliative, mais aussi des lésions cutanées, un œdème de la face, de la fièvre, des anomalies du bilan sanguin telles qu’une thrombocytopénie et une éosinophilie, et atteinte d’un organe unique ou multiviscérale (du foie et des reins incluant une néphrite tubulo-interstitielle) (voir rubrique 4.4).
Les crises de goutte ont fréquemment été observées peu après le début du traitement et au cours des premiers mois. Par la suite, la fréquence des crises de goutte diminue dans le temps. Une prophylaxie des crises de goutte est recommandée (voir rubriques 4.2 et 4.4).
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : www.signalement-sante.gouv.fr.
Le traitement d’un surdosage doit être symptomatique et comporter des mesures de soutien.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Mécanisme d’action
L’acide urique est le produit final du métabolisme des purines chez l’homme et résulte de la cascade hypoxanthine → xanthine → acide urique. Ces deux étapes sont catalysées par la xanthine oxydase (XO). Le fébuxostat est un dérivé 2-arylthiazole qui exerce son effet thérapeutique de diminution de l’uricémie en inhibant sélectivement la XO. Le fébuxostat est un inhibiteur non purinique puissant et sélectif de la XO (NP-SIXO). In vitro, sa constante d’inhibition Ki est inférieure à une nanomole. Le fébuxostat inhibe de façon puissante les formes oxydée et réduite de la XO. Aux concentrations thérapeutiques, le fébuxostat n’inhibe pas les autres enzymes intervenant dans le métabolisme des purines ou des pyrimidines (guanine désaminase, hypoxanthine guanine phosphoribosyltransférase, orotate phosphoribosyltransférase, orotidine monophosphate décarboxylase ou purine nucléoside phosphorylase).
Efficacité et sécurité clinique
L’efficacité du fébuxostat a été démontrée au cours de trois études pivots de phase III (les deux études pivots APEX et FACT et l’étude additionnelle CONFIRMS décrites ci-dessous) menées chez 4101 patients présentant une hyperuricémie et une goutte. Dans chacune des études pivots de phase III, le fébuxostat a démontré sa supériorité vis à vis de l’allopurinol pour diminuer et maintenir l’uricémie. Le critère principal d’efficacité au cours des études APEX et FACT était la proportion des patients présentant une uricémie < 6,0 mg/dL (357 µmol/L) au cours des 3 dernières mesures mensuelles. Au cours de l’étude additionnelle de phase III CONFIRMS, dont les résultats ont été obtenus après l’octroi de l’autorisation de mise sur le marché de fébuxostat, le critère principal d’efficacité était la proportion de patients présentant une uricémie < 6,0 mg/dL à la dernière visite. Aucun patient ayant reçu une greffe d’organe n’a été inclus dans ces études (voir rubrique 4.2).
Étude APEX : L’étude APEX (Allopurinol and Placebo-Controlled Efficacy Study of Febuxostat) est une étude de phase 3 multicentrique randomisée, menée en double insu, d’une durée de 28 semaines contrôlée contre placebo et allopurinol. Mille soixante-douze (1072) patients ont été randomisés dans les groupes suivants : placebo (n=134), 80 mg de fébuxostat une fois par jour (n=267), 120 mg de fébuxostat une fois par jour (n=269), 240 mg de fébuxostat une fois par jour (n=134) ou allopurinol (300 mg une fois par jour [n=258] chez les patients dont la créatinémie initiale était ≤ 1,5 mg/dL ou 100 mg une fois par jour [n=10] chez ceux dont la créatinémie initiale était > 1,5 mg/dL et ≤ 2,0 mg/dL). La dose de 240 mg de fébuxostat (deux fois la plus forte dose recommandée) a été étudiée pour évaluer la tolérance.
L'étude APEX a démontré la supériorité statistiquement significative du fébuxostat 80 mg une fois par jour et du fébuxostat T ARROW 120 mg une fois par jour par rapport à l’allopurinol administré aux doses conventionnelles de 300 mg (n = 258) /100 mg (n = 10) sur la diminution de l’uricémie en dessous du seuil de 6 mg/dL (357 µmol/L) (voir tableau 2 et figure 1).
Étude FACT : L'étude FACT (Febuxostat Allopurinol Controlled Trial) est une étude de phase 3 multicentrique randomisée, menée en double insu, d’une durée de 52 semaines, contrôlée contre allopurinol. Sept cent soixante (760) patients ont été randomisés dans les groupes suivants : Fébuxostat 80 mg une fois par jour (n = 256), Fébuxostat 120 mg une fois par jour (n = 251) et allopurinol 300 mg une fois par jour (n = 253).
L'étude FACT a montré la supériorité statistiquement significative de Fébuxostat 80 mg une fois par jour et Fébuxostat 120 mg une fois par jour par rapport à l’allopurinol administré à la dose conventionnelle de 300 mg sur la réduction et le maintien de l’uricémie au-dessous du seuil de 6 mg/dL (357 µmol/L).
Le tableau 2 résume les résultats sur le critère principal d’efficacité.
Tableau 2 : Proportion des patients présentant une uricémie < 6,0 mg/dL (357 µmol/L) Au cours des trois dernières visites mensuelles
|
Étude |
Fébuxostat 80 mg 1x/jour |
Fébuxostat 120 mg 1x/jour |
Allopurinol 300/ 100 mg 1x/jour1 |
|
APEX (28 semaines) |
48% * (n=262) |
65% *, # (n=269) |
22% (n=268) |
|
FACT (52 semaines) |
53%* (n=255) |
62%* (n=250) |
21% (n=251) |
|
Résultats regroupés |
51%* (n=517) |
63%*, # (n=519) |
22% (n=519) |
|
1Les résultats observés chez les sujets recevant 100 mg une fois par jour (n = 10, créatininémie > 1,5 et ≤ 2,0 mg/dL) ou 300 mg une fois par jour (n=509) ont été regroupés pour les analyses. * p < 0,001 vs allopurinol, # p < 0,001 vs 80 mg |
|||
La diminution de l'uricémie sous l’effet du féboxustat a été rapide et persistante. Une réduction de l’uricémie sous le seuil de 6,0 mg/dL (357 µmol/L) a été notée dès la visite en semaine 2 et s’est maintenue pendant toute la durée du traitement. La figure 1 présente l’évolution de l’uricémie moyenne au cours du temps dans chaque groupe de traitement au cours des deux études pivots de phase 3.
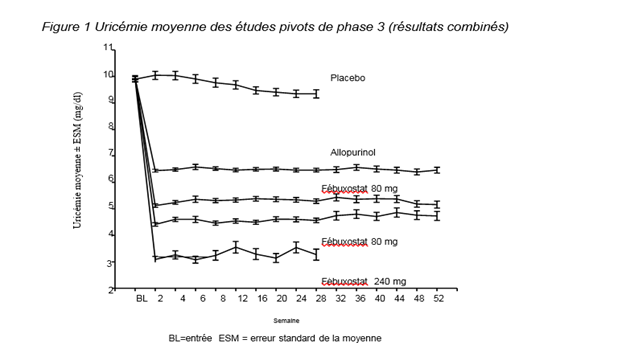
Note : 509 patients ont reçu l’allopurinol à raison de 300 mg 1x/jour ; 10 patients dont la créatininémie était > 1,5 et ≤ 2.0 mg/dL ont reçu 100 mg 1x/jour (10 patients sur 268 dans l’étude APEX).
La dose de 240 mg a été utilisée pour évaluer la tolérance du fébuxostat à une dose deux fois supérieure à la dose maximale recommandée).
Etude CONFIRMS : l’étude CONFIRMS est une étude de phase 3, randomisée, contrôlée, d’une durée de 26 semaines dont l’objectif était d’évaluer la tolérance et l’efficacité du fébuxostat 40 et 80 mg comparativement à l’allopurinol 300 ou 200 mg chez des patients atteints de goutte et présentant une hyperuricémie. 2 269 patients ont été randomisés : groupe fébuxostat 40 mg une fois par jour (n = 757), groupe fébuxostat 80 mg une fois par jour (n = 756), groupe allopurinol 300/200 mg une fois par jour (n = 756). Au moins 65 % des patients avaient une insuffisance rénale légère (clairance de la créatinine comprise entre 30 et 89 ml/min). Une prophylaxie des crises de goutte était obligatoire pendant les 26 semaines de traitement.
La proportion de patients avec une uricémie < 6 mg/dL (357 µmol/L) à la dernière visite était de 45 % dans le groupe fébuxostat 40 mg, 67 % dans le groupe fébuxostat 80 mg et 42 % dans le groupe allopurinol 300/200 mg.
Critère principal dans le sous-groupe des patients insuffisants rénaux
L’étude APEX a évalué l’efficacité chez 40 patients insuffisants rénaux (définie par une créatininémie initiale > 1,5 mg/dL et ≤ 2,0 mg/dL). Chez les insuffisants rénaux randomisés dans le groupe allopurinol, la dose a été limitée à 100 mg une fois par jour. Le critère principal d’efficacité a été atteint sous fébuxostat chez 44 % (80 mg une fois par jour), 45 % (120 mg une fois par jour) et 60 % (240 mg une fois par jour) des patients contre 0 % des patients inclus dans le groupe allopurinol 100 mg une fois par jour et dans le groupe placebo.
La diminution de l’uricémie en pourcentage n’a pas différé de façon cliniquement significative en fonction de l’état de la fonction rénale (58 % dans le groupe fonction rénale normale et 55 % dans le groupe dysfonction rénale sévère).
Une analyse, définie de façon prospective dans l’étude CONFIRMS, effectuée chez les patients atteints de goutte présentant une insuffisance rénale légère à modérée (65 % des patients étudiés) a montré que le fébuxostat était significativement plus efficace que l’allopurinol 300/200 mg pour abaisser l’uricémie en deçà de 6 mg/dL.
Critère principal dans le sous-groupe des patients présentant une uricémie ≥ 10 mg/dL
L’uricémie initiale était ≥ 10 mg/dL chez environ 40 % des patients inclus dans les études APEX et FACT (considérées simultanément). Dans ce sous-groupe, le critère principal d’efficacité (uricémie < 6,0 mg/dL aux 3 dernières visites) a été atteint sous fébuxostat chez 41 % (80 mg une fois par jour), 48 % (120 mg une fois par jour) et 66 % (240 mg une fois par jour) des patients contre 9 % des patients inclus dans le groupe allopurinol 300 mg/100 mg une fois par jour et 0 % dans le groupe placebo.
Au cours de l’étude CONFIRMS, la proportion de patients ayant atteint le critère principal d’efficacité (uricémie < 6 mg/dL à la dernière visite) parmi ceux ayant une uricémie initiale ≥ 10 mg/dL était de 27 % (66/249) chez les patients traités par fébuxostat 40 mg une fois par jour, 49 % (125/254) chez les patients traités par fébuxostat 80 mg une fois par jour et 31 % (72/230) chez les patients traités par allopurinol 300/200 mg.
Critères cliniques : proportion de patients ayant nécessité un traitement de la crise de goutte
Etude APEX : au cours de la période de prophylaxie de 8 semaines, une proportion plus importante de sujets du groupe fébuxostat 120 mg (36 %) a nécessité un traitement de la crise de goutte comparativement aux groupes fébuxostat 80 mg (22 %), allopurinol 300 mg (23 %) et placebo (20 %).
Les crises ont augmenté après la période de prophylaxie puis ont diminué graduellement au cours du temps. Entre 46 % et 55 % des sujets ont reçu un traitement de la crise de goutte de la semaine 8 à la semaine 28. Les crises de goutte survenues durant les 4 dernières semaines de l’étude (semaine 24 – semaine 28) ont été observées chez 15 % des sujets du groupe fébuxostat 80/120 mg, 14 % des sujets du groupe allopurinol 300 mg et 20 % des sujets du groupe placebo.
Etude FACT : au cours de la période de prophylaxie de 8 semaines, une proportion plus importante de sujets du groupe fébuxostat 120 mg (36 %) a nécessité un traitement de la crise de goutte comparativement aux groupes fébuxostat 80 mg (22 %) et allopurinol 300 mg (21 %). Après la période de prophylaxie de 8 semaines, l’incidence des crises a augmenté puis a graduellement diminué au cours du temps (64 % et 70 % des sujets ont reçu un traitement de la crise de goutte de la semaine 8 à la semaine 52). Les crises de goutte survenues durant les 4 dernières semaines de l’étude (semaine 49 – semaine 52) ont été observées chez 6 à 8 % des sujets du groupe fébuxostat 80/120 mg et 11 % des sujets du groupe allopurinol 300 mg.
La proportion des sujets ayant nécessité un traitement de la crise de goutte (études APEX et FACT) a été numériquement plus faible dans les groupes où l’uricémie moyenne après l’entrée dans l’étude avait été < 6,0 mg/dL, < 5,0 mg/dL ou < 4,0 mg/dL que dans le groupe où elle avait été ≥ 6,0 mg/dL au cours des 32 dernières semaines de traitement (intervalles semaine 20 – semaine 24 à semaines 49 – 52).
Au cours de l’étude CONFIRMS, les proportions de patients ayant nécessité un traitement de la crisede goutte (du 1er jour au 6ème mois) étaient de 31 % et 25 % respectivement dans le groupe fébuxostat 80 mg et le groupe allopurinol. Aucune différence n’a été observée entre la proportion de patients ayant nécessité un traitement de la crise de goutte entre le groupe fébuxostat 80 mg et le groupe fébuxostat 40 mg.
Etudes d’extension en ouvert à long terme
Etude EXCEL (C02-021) : l’étude EXCEL était une étude d’extension de phase 3, d’une durée de 3 ans, effectuée en ouvert, multicentrique, randomisée, contrôlée contre allopurinol, évaluant la tolérance chez les patients qui avaient terminé les études pivots de phase 3 (APEX ou FACT). Au total 1 086 patients ont été inclus : groupe fébuxostat 80 mg une fois par jour (n = 649), groupefébuxostat 120 mg une fois par jour (n = 292) et groupe allopurinol 300/100 mg une fois par jour (n= 145). Environ 69 % des patients n’ont pas nécessité de modification de leur traitement pour parvenir à un traitement final stable. Les patients ayant 3 mesures d’uricémie consécutives > 6 mg/dL ont étésortis de l’étude. Les niveaux d’uricémie se sont maintenus au cours du temps (91 % et 93 % respectivement des patients traités par fébuxostat 80 mg et 120 mg avaient une uricémie < 6 mg/dL à 36 mois).
Les données recueillies pendant 3 ans ont montré une diminution de l’incidence des crises de goutte, un traitement pour une crise de goutte s’étant avéré nécessaire chez moins de 4 % des patients (plus de 96 % des patients n’ont pas été traités pour une crise de goutte) entre les 16ème et 24ème mois et entre les 30ème et 36ème mois.
Respectivement 46 et 38 % des patients ayant un traitement final stable par fébuxostat 80 ou 120 mg une fois par jour ont eu une résolution complète du premier tophus palpable entre la visite initiale et la dernière visite.
L’étude TMX-01-005 (FOCUS) était une étude d’extension de phase 2 d’une durée de 5 ans, en ouvert, multicentrique, évaluant la tolérance chez les patients qui avaient terminé les 4 semaines de traitement par fébuxostat en double aveugle de l’étude de détermination de doses TMX-00-004. 116 patients ont été inclus et ont été traités par fébuxostat 80 mg une fois par jour. 62 % des patients n’ont pas nécessité d’ajustement de la posologie pour maintenir une uricémie < 6 mg/dL et 38 % des patients ont nécessité une adaptation de la posologie avant d’atteindre un traitement final stable.
La proportion de patients avec une uricémie < 6 mg/dL (357 µmol/L) à la dernière visite était supérieure à 80 % (de 81 à 100 %) pour chacune des doses de fébuxostat.
Au cours des études cliniques de phase 3, de légères anomalies du bilan hépatique ont été enregistrées chez des patients (5,0 %) traités par le fébuxostat. Ce pourcentage a été similaire à celui rapporté avec l’allopurinol (4,2 %) (voir rubrique 4.4). Au cours des études d’extension ouvertes à long terme, une augmentation du taux de TSH (> 5,5 µUI/mL) a été constatée chez des patients traités au long cours par le fébuxostat (5,5 %) et par l’allopurinol (5,8 %) (voir rubrique 4.4).
Étude après commercialisation à long terme
L’étude CARES est une étude clinique multicentrique, randomisée, en double aveugle, de non infériorité qui a comparé l’impact cardiovasculaire du fébuxostat versus allopurinol chez des patients atteints de goutte et ayant des antécédents de maladie cardiovasculaire sévère incluant infarctus du myocarde, hospitalisation pour angor instable, procédure de revascularisation coronaire ou cérébrale, AVC, hospitalisation pour accident ischémique transitoire, maladie vasculaire périphérique, ou diabète avec complications microvasculaires ou macrovasculaires. Pour atteindre une uricémie inférieure à 6 mg/dL, la dose de fébuxostat a été ajustée de 40 mg à 80 mg (sans prendre en compte la fonction rénale) et la dose d’allopurinol a été ajustée par paliers de 100 mg de 300 à 600 mg chez les patients ayant une fonction rénale normale ou une insuffisance rénale légère et de 200 mg à 400 mg chez les patients avec une insuffisance rénale modérée.
Le critère de jugement principal de l’étude CARES a été la première survenue du critère composite « Événements cardiovasculaires majeurs » (MACE) regroupant infarctus non mortels, AVC non mortels, décès cardiovasculaires, angor instable avec revascularisation coronarienne en urgence. Les critères de jugement (principal et secondaires) ont été analysés en intention de traiter (ITT) en incluant tous les sujets ayant été randomisés et ayant reçu au moins une dose du médicament en double aveugle.
Globalement, 56,6 % des patients ont interrompu leur traitement prématurément et 45% des patients ne se sont pas présentés à l’ensemble des visites de l’essai clinique.
Au total, 6190 patients ont été suivis avec une durée médiane de 32 mois, et la durée médiane d’exposition a été de 728 jours pour les patients dans le groupe fébuxostat (n = 3098) et de 719 jours dans le groupe allopurinol (n = 3092).
Le critère de jugement principal MACE est survenu à un taux semblable dans les groupes fébuxostat et allopurinol (respectivement 10,8 % vs 10,4 % ; HR 1,03 ; intervalle de confiance bilatéral à 95% [IC] 0,89-1,21).
Dans l’analyse des composantes individuelles du critère MACE, le taux de décès cardiovasculaire a été supérieur avec le fébuxostat qu’avec l’allopurinol (4,3 % vs 3,2 % ; HR 1,34 ; IC 95% 1,03-1,73). Les taux des autres évènements MACE ont été similaires dans les groupes fébuxostat et allopurinol, c’est-à-dire pour les infarctus du myocarde non mortels (3,6 % vs 3,8 % ; HR 0,93 ; IC 95% 0,72-1,21), AVC non mortels (2,3 % vs 2,3 % ; HR 1,01 ; IC 95% 0,73-1,41) et la revascularisation en urgence suite à un angor instable (1,6 % vs 1,8 % ; HR 0,86 ; IC 95% 0,59-1,26). Le taux de décès toutes causes confondues a été aussi supérieur avec le fébuxostat versus allopurinol (7,8 % vs 6,4 % ; HR 1,22 ; IC 95% 1,01-1,47), du fait principalement du taux supérieur de décès cardiovasculaires dans le groupe fébuxostat (voir rubrique 4.4).
Les taux d’hospitalisations attribuées à une insuffisance cardiaque, d’admissions hospitalières pour arythmie non associées à une ischémie, d’évènements thromboemboliques veineux et d’hospitalisations pour accident ischémique transitoire ont été comparables avec le fébuxostat et l’allopurinol.
L’étude FAST est une étude prospective, randomisée, en ouvert, en aveugle, qui a comparé le profil de tolérance cardiovasculaire (CV) du fébuxostat versus allopurinol chez des patients atteints d’hyperuricémie chronique (dans les cas où un dépôt d’urate s’est déjà produit) et avec des facteurs de risque CV (c’est-à-dire des patients âgés de 60 ans ou plus et présentant au moins un autre facteur de risque CV). Les patients éligibles ont reçu un traitement par allopurinol avant leur randomisation et des ajustements de dose ont été effectués si besoin, selon le jugement clinique, les recommandations EULAR et la posologie approuvée par l’AMM. A la fin de la phase d'introduction de l'allopurinol, les patients avec un taux sérique d'acide urique < 0,36 mmol/L (< 6 mg/dL) ou recevant la dose maximale tolérée ou la dose maximale autorisée d'allopurinol ont été randomisés dans un rapport 1:1 afin de recevoir un traitement par fébuxostat, ou par allopurinol. Le critère de jugement principal de l’étude FAST était le délai d’apparition de la première survenue d’évènements inclus dans le critère composite « Antiplatelet Trialists Collaborative » (APTC) qui incluait : i) hospitalisation pour infarctus du myocarde non-fatal/marqueur biologique positif du syndrome coronaire aigu (SCA) ; ii) AVC non fatal ; iii) mortalité due à un évènement CV. L’analyse primaire s’est basée sur une approche « en traitement reçu » (on-treatment (OT)).
Au total, 6128 patients ont été randomisés, 3063 avec le fébuxostat et 3065 avec l’allopurinol.
Dans l’analyse primaire OT, le fébuxostat était non inférieur à l’allopurinol sur l’incidence du critère de jugement principal, qui est survenu chez 172 patients (1,72/100 patients-années) sous fébuxostat comparé au 241 patients (2,05/100 patients-années) sous allopurinol, avec un HR ajusté de 0,85 (IC 95 % : 0,70-1,03), p < 0,001. L’analyse OT du critère de jugement principal dans le sous-groupe des patients avec des antécédents d’IDM, d’AVC ou de SCA n’a pas montré de différence significative entre les groupes de traitement : il y a eu 65 (9,5 %) patients avec des évènements dans le groupe fébuxostat et 83 (11,8 %) patients avec des évènements dans le groupe allopurinol ; HR ajusté 1,02 (IC 95 % : 0,74-1,42) ; p = 0,202.
Le traitement par le fébuxostat n’a pas été associé à une augmentation de la mortalité CV ou de la mortalité toute cause, globalement ou dans les sous-groupes de patients avec un antécédent d’IDM, d’AVC ou de SCA. Au final, il y a eu moins de décès dans le groupe fébuxostat (62 décès d’origine CV et 108 décès toute cause), que dans le groupe allopurinol (82 décès d’origine CV et 174 décès toute cause).
Il y a eu une plus grande réduction du taux d’acide urique sous traitement avec fébuxostat comparé au traitement avec allopurinol.
5.2. Propriétés pharmacocinétiques
Des analyses pharmacocinétiques/pharmacodynamiques de population ont été menées chez 211 patients présentant une hyperuricémie et une goutte, qui ont été traités par fébuxostat 40 à 240 mg une fois par jour. En règle générale, les paramètres pharmacocinétiques du fébuxostat estimés par ces analyses ont été similaires à ceux déterminés chez les sujets sains, indiquant que ces derniers sont représentatifs pour l’évaluation pharmacocinétique/ pharmacodynamique chez les patients atteints de goutte.
Absorption
L’absorption du fébuxostat est rapide (tmax = 1,0 – 1 ,5 h) et élevée (au moins 84 %). Après des doses orales uniques ou répétées de 80 et 120 mg une fois par jour, la Cmax est respectivement d’environ 2,8-3,2 µg/mL et 5,0-5,3 µg/mL. La biodisponibilité absolue de la formulation comprimé du fébuxostat n’a pas été étudiée.
A la suite de doses orales répétées de 80 mg une fois par jour ou d’une dose unique de 120 mg avec un repas riche en lipide, la Cmax a diminué de respectivement 49 % et 38 % et l’ASC de 18 % et 16 %. Aucune modification cliniquement significative du pourcentage de diminution de l’uricémie n’a été cependant observée quand ce paramètre a été mesuré (doses répétées de 80 mg). Fébuxostat peut donc être pris conjointement ou non avec une prise alimentaire.
Distribution
Le volume apparent de distribution à l’état d’équilibre (Vss/F) du fébuxostat est de 29 à 75 L après des doses orales de 10 à 300 mg. La liaison du fébuxostat aux protéines plasmatiques est d’environ 99,2 % (principalement à l’albumine) et est constante avec les concentrations obtenues avec les doses de 80 et 120 mg. La liaison des métabolites actifs aux protéines plasmatiques est d’environ 82 % à 91 %.
Biotransformation
Le fébuxostat est fortement métabolisé par conjugaison via le système enzymatique diphosphate glucuronosyltransférase (UDPGT) et par oxydation via le cytochrome P450 (CYP). Quatre métabolites hydroxylés pharmacologiquement actifs ont été identifiés, dont trois ont été décelés dans le plasma chez l'homme. Des études in vitro sur microsomes hépatiques humains ont montré que ces métabolites oxydatifs étaient principalement formés par CYP1A1, CYP1A2, CYP2C8 ou CYP2C9 et que le glycuronide du fébuxostat était principalement formé par UGT 1A1, 1A8 et 1A9.
Élimination
Le fébuxostat est éliminé par voies hépatique et rénale. Après administration par voie orale d’une dose de 80 mg de fébuxostat marqué au 14C, environ 49 % de la dose a été retrouvée dans l’urine sous forme de fébuxostat inchangé (3 %), d’acyl glycuronide de la substance active (30 %), de ses métabolites oxydatifs connus et de leurs dérivés conjugués (13 %) et d’autres métabolites inconnus (3 %). En dehors de l’excrétion urinaire, près de 45% de la dose a été retrouvée dans les fèces sous forme de fébuxostat inchangé (12 %), d’acyl glycuronide de la substance active (1 %), de ses métabolites oxydatifs connus et de leurs dérivés conjugués (25 %) et d’autres métabolites inconnus (7 %).
Insuffisance rénale
Après administration de doses répétées de 80 mg de fébuxostat, la Cmax du fébuxostat n’est pas différente entre les patients présentant une insuffisance rénale légère, modérée ou sévère par rapport à des sujets à fonction rénale normale. L’ASC moyenne totale du fébuxostat a été environ 1,8 fois plus élevée chez les patients présentant une dysfonction rénale sévère que chez les sujets à fonction rénale normale (13,2 µg.h/mL contre 7,5 µg.h/mL). La Cmax et l’ASC des métabolites actifs ont été respectivement deux et quatre fois plus élevées. Aucune adaptation de la posologie n’est cependant nécessaire chez les patients présentant une insuffisance rénale légère à modérée.
Insuffisance hépatique
Après administration de doses répétées de 80 mg de Fébuxostat, la Cmax et l’ASC du fébuxostat et de ses métabolites ne sont significativement différents entre les patients présentant une insuffisance hépatique légère (classe A de Child-Pugh) ou modérée (classe B de Child-Pugh) par rapport à des sujets à fonction hépatique normale. Aucune étude n’a été menée chez des patients présentant une insuffisance hépatique sévère (classe C de Child-Pugh).
Age
Après administration répétée de Fébuxostat par voie orale, aucune différence significative de l’ASC du fébuxostat n’a été observée entre des sujets âgés et des sujets sains plus jeunes.
Sexe
Après administration répétée de Fébuxostat par voie orale, la Cmax et l’ASC du fébuxostat ont été plus élevées de respectivement 24 % et 12 % chez les femmes que chez les hommes. La Cmax et l’ASC corrigées en fonction du poids ont été cependant similaires entre les sujets des deux sexes. Aucune adaptation de la dose en fonction du sexe n’est nécessaire.
5.3. Données de sécurité préclinique
La modélisation pharmacocinétique et les données de simulation chez le rat suggèrent que, en cas de co-administration avec le fébuxostat, la dose clinique de mercaptopurine/azathioprine doit être réduite à 20 % ou moins de la dose préalablement prescrite afin d'éviter d'éventuels effets hématologiques (voir les rubriques 4.4 et 4.5).
Cancérogenèse, mutagenèse, altération de la fertilité
Chez le rat mâle, une augmentation statistiquement significative des tumeurs de la vessie (papillomes et carcinomes à cellules transitionnelles) n’a été observée qu’en association à des calculs de xanthine dans le groupe recevant une dose élevée (environ 11 fois l’exposition humaine). Aucune augmentation significative d’un autre type de tumeur n'a été observée chez la souris et le rat mâle ou femelle. Ces observations sont considérées comme une conséquence d’une composition de l’urine et d’un métabolisme des purines spécifiques à l’espèce et comme dépourvues de signification en clinique.
Une batterie standard de tests de génotoxicité n’a révélé aucun effet génotoxique biologiquement pertinent du fébuxostat.
Le fébuxostat à des doses orales allant jusqu’à 48 mg/jour n’a montré aucun effet sur la fertilité et la capacité de reproduction chez le rat mâle ou femelle.
Aucun signe d’altération de la fertilité, d’effet tératogène ou d’effet délétère sur le fœtus lié au fébuxostat n’a été observé. Une toxicité maternelle a été observée aux doses élevées, accompagnée d'une réduction de l’indice de sevrage et du développement des petits chez le rat à une exposition d’environ 4,3 fois celle observée chez l’homme. Des études de tératogenèse menées chez la rate gestante à une exposition équivalente à environ 4,3 fois l'exposition humaine et chez la lapine gestante à une exposition d’environ 13 fois celle-ci n’ont révélé aucun effet tératogène.
Noyau du comprimé : lactose monohydraté, cellulose microcristalline (grade 101), croscarmellose sodique, hydroxypropylcellulose, cellulose microcristalline (grade 102), silice colloïdale anhydre, stéarate de magnésium.
Pelliculage du comprimé : alcool polyvinylique, dioxyde de titane (E 171), macrogol 3350, talc, oxyde de fer jaune (E 172).
3 ans.
6.4. Précautions particulières de conservation
Ce médicament ne nécessite pas de précautions particulières de conservation.
6.5. Nature et contenu de l'emballage extérieur
FÉBUXOSTAT ARROW 80 mg, comprimé pelliculé est disponible sous plaquettes transparentes (PVC/Aluminium ou PVC/PE/PVDC/Aluminium).
FÉBUXOSTAT ARROW 80 mg, comprimé pelliculé est présenté en boîtes de 14, 28, 30, 42, 56, 84 et 98 comprimés pelliculés.
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
26 AVENUE TONY GARNIER
69007 LYON
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 301 709 0 8 : Comprimés pelliculés sous plaquettes transparentes (PVC/Aluminium) ; boîte de 28.
· 34009 301 709 1 5 : Comprimés pelliculés sous plaquettes transparentes (PVC/PE/PVDC/Aluminium) ; boîte de 28.
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I
ANSM - Mis à jour le : 02/05/2022
FÉBUXOSTAT ARROW 80 mg, comprimé pelliculé
Fébuxostat
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin ou votre pharmacien.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que FÉBUXOSTAT ARROW 80 mg, comprimé pelliculé et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant de prendre FÉBUXOSTAT ARROW 80 mg, comprimé pelliculé ?
3. Comment prendre FÉBUXOSTAT ARROW 80 mg, comprimé pelliculé ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver FÉBUXOSTAT ARROW 80 mg, comprimé pelliculé ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE FÉBUXOSTAT ARROW 80 mg, comprimé pelliculé ET DANS QUELS CAS EST-IL UTILISE ?
FÉBUXOSTAT ARROW agit en réduisant le taux d’acide urique dans le sang (uricémie). Le maintien de l’uricémie à un niveau bas, grâce à la prise quotidienne de FÉBUXOSTAT ARROW, permet d’arrêter l'accumulation de cristaux et de réduire progressivement les symptômes. Le maintien d’une uricémie suffisamment basse pendant une durée suffisamment longue peut également permettre une diminution de la taille des tophi.
FEBUXOSTAT ARROW comprimé à 120 mg est aussi utilisé pour traiter et éviter une élévation du taux sanguin d’acide urique, qui peut apparaître quand on commence à recevoir une chimiothérapie pour traiter les cancers du sang.
Quand on reçoit une chimiothérapie, les cellules cancéreuses sont détruites, et par conséquent le taux d’acide urique augmente dans le sang, à moins que l’on empêche la formation d’acide urique.
FÉBUXOSTAT ARROW est indiqué chez l’adulte.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT DE PRENDRE FÉBUXOSTAT ARROW 80 mg, comprimé pelliculé ?
Ne prenez jamais FÉBUXOSTAT ARROW 80 mg, comprimé pelliculé :
· si vous êtes allergique au fébuxostat ou à l’un des autres composants contenus dans ce médicament, mentionnés dans la rubrique 6.
Avertissements et précautions
Adressez-vous à votre médecin avant de prendre FÉBUXOSTAT ARROW 80 mg, comprimé pelliculé :
· si vous avez ou avez eu une insuffisance cardiaque, des problèmes cardiaques ou un AVC ;
· si vous avez ou avez eu une maladie des reins et/ou une réaction allergique grave à l’allopurinol (un médicament utilisé pour le traitement de la goutte) ;
· si vous avez ou avez eu une maladie du foie ou une anomalie des paramètres de la fonction hépatique ;
· si vous êtes traité(e) pour une uricémie élevée due à un syndrome de Lesch-Nyhan (affection héréditaire rare, au cours de laquelle le taux d’acide urique dans le sang est trop élevé) ;
· si vous avez des problèmes thyroïdiens.
En cas de réaction allergique à FÉBUXOSTAT ARROW, arrêtez de prendre ce médicament (voir rubrique 4). Les symptômes d’une réaction allergique au fébuxostat peuvent être :
· une éruption cutanée pouvant être des formes graves (avec des cloques, des nodules, des démangeaisons, une éruption exfoliative), des démangeaisons ;
· un gonflement des membres ou de la face ;
· une difficulté à respirer ;
· de la fièvre avec un gonflement des ganglions lymphatiques ;
· mais également des manifestations allergiques graves, potentiellement fatales par arrêt cardiaque ou circulatoire.
Votre médecin peut décider l’arrêt définitif de votre traitement par FÉBUXOSTAT ARROW.
De rares cas d’éruption cutanée pouvant être fatals (syndrome de Stevens-Johnson) ont été observés lors de l’utilisation de FÉBUXOSTAT ARROW, apparaissant au début au niveau du tronc sous forme de boutons rougeâtres localisés ou des plaques circulaires avec, en leurs centres, des cloques. Des ulcères dans la bouche, la gorge, le nez, au niveau des parties génitales et une conjonctivite (yeux rouges et gonflés) peuvent également apparaître. L’éruption cutanée peut évoluer sur l’ensemble du corps vers la formation de cloques ou un décollement de la peau.
Si vous développez un syndrome de Stevens-Johnson lors de l’utilisation de FÉBUXOSTAT ARROW, votre traitement par fébuxostat ne devra jamais être réinitié. Si vous présentez une éruption cutanée ou des symptômes cutanés, vous devez prendre immédiatement l’avis d’un médecin et lui dire que vous prenez ce médicament.
Si vous avez actuellement une crise de goutte (survenue subite de douleurs intenses, d’une sensibilité, d’une rougeur, d’une chaleur et d'un gonflement d'une articulation), attendez que la crise de goutte disparaisse totalement avant de débuter le traitement par FÉBUXOSTAT ARROW.
Chez certaines personnes, des crises de goutte peuvent apparaître au début d’un traitement par certains médicaments qui diminuent le taux d’acide urique. Ces crises n’apparaissent pas chez toutes les personnes traitées, mais peuvent survenir lors de la prise de FÉBUXOSTAT ARROW, particulièrement au cours des premières semaines ou des premiers mois qui suivent le début du traitement. Il est important de continuer à prendre FÉBUXOSTAT ARROW même si vous avez une crise de goutte, car ce médicament continuera à agir pour diminuer l’uricémie. Les crises de goutte deviendront moins fréquentes et moins douloureuses au cours du temps si vous continuez à prendre FÉBUXOSTAT ARROW tous les jours.
Votre médecin prescrira souvent d’autres médicaments, s’ils sont nécessaires, afin de prévenir ou de traiter les symptômes d'une crise de goutte (par exemple douleurs et gonflement d’une articulation).
Chez les patients ayant un taux très élevé d’acide urique (par exemple, sous chimiothérapie anticancéreuse), le traitement par des médicaments diminuant l’acide urique peut conduire à l’accumulation de xanthine dans les voies urinaires, avec la possible formation de calculs, même si cela n’a pas été observé chez les patients traités avec FÉBUXOSTAT ARROW pour un Syndrome de Lyse Tumorale.
Votre médecin pourra vous prescrire des analyses de sang afin de vérifier que votre foie fonctionne normalement.
Enfants et adolescents
Ne jamais donner ce médicament aux enfants de moins de 18 ans étant donné que la sécurité et l’efficacité de ce médicament dans cette population n’ont pas été établies.
Autres médicaments et FÉBUXOSTAT ARROW 80 mg, comprimé pelliculé
Informez votre médecin ou votre pharmacien si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament, y compris un médicament obtenu sans ordonnance.
Il est particulièrement important de prévenir votre médecin ou votre pharmacien si vous prenez un médicament contenant l’une des substances suivantes, car il peut interagir avec FÉBUXOSTAT ARROW, et votre médecin peut vouloir envisager les mesures nécessaires :
· mercaptopurine (utilisée dans le traitement du cancer) ;
· azathioprine (utilisée pour réduire les réponses immunitaires) ;
· théophylline (utiliser pour traiter l’asthme).
On ne sait pas si FÉBUXOSTAT ARROW peut être nocif pour l’enfant à naître. FÉBUXOSTAT ARROW ne doit pas être pris pendant la grossesse. On ne sait pas si FÉBUXOSTAT ARROW passe dans le lait maternel. Vous ne devez pas prendre FÉBUXOSTAT ARROW si vous allaitez ou si vous prévoyez de le faire.
Si vous êtes enceinte ou que vous allaitez, si vous pensez être enceinte ou planifiez une grossesse, demandez conseil à votre médecin ou votre pharmacien avant de prendre ce médicament.
Conduite de véhicules et utilisation de machines
Vous pouvez ressentir des étourdissements, une somnolence, une vision floue ou des sensations d’engourdissement ou de picotements lors du traitement et que dans ce cas, vous ne devez pas conduire des véhicules ou utiliser des machines.
FÉBUXOSTAT ARROW 80 mg, comprimé pelliculé contient du lactose et du sodium
Si vous êtes intolérant à certains sucres, parlez-en à votre médecin avant de prendre ce médicament.
Ce médicament contient moins de 1 mmol (23 mg) de sodium par comprimé pelliculé, c’est-à-dire qu’il est essentiellement « sans sodium ».
3. COMMENT PRENDRE FÉBUXOSTAT ARROW 80 mg, comprimé pelliculé ?
Veillez à toujours prendre ce médicament en suivant exactement les indications de votre médecin. Vérifiez auprès de votre médecin ou de votre pharmacien en cas de doute.
· La posologie habituelle est d’un comprimé par jour.
· Les comprimés doivent être pris par voie orale et peuvent être pris avec ou sans aliments.
Goutte
FÉBUXOSTAT ARROW est disponible sous forme de comprimés dosés à 80 mg ou 120 mg. Votre médecin vous a prescrit le dosage le mieux adapté à votre cas.
Continuez à prendre FÉBUXOSTAT ARROW tous les jours même si vous n’avez pas de crise de goutte.
Prévention et traitement du taux élevé d’acide urique chez les patients sous chimiothérapie anticancéreuse
FÉBUXOSTAT ARROW est disponible sous forme de comprimés à 120 mg.
Commencer à prendre FÉBUXOSTAT ARROW deux jours avant la chimiothérapie et poursuivre selon les conseils de votre médecin. Habituellement, le traitement est de courte durée.
Si vous avez pris plus de FÉBUXOSTAT ARROW 80 mg, comprimé pelliculé que vous n’auriez dû
Si vous avez pris plus de FÉBUXOSTAT ARROW que vous n’auriez dû, contactez votre médecin ou le service des urgences le plus proche.
Si vous oubliez de prendre FÉBUXOSTAT ARROW 80 mg, comprimé pelliculé
Si vous oubliez de prendre une dose de FÉBUXOSTAT ARROW, prenez-là dès que vous vous apercevez de votre oubli, sauf s’il est presque le moment de la prise suivante. Dans ce cas, ne prenez pas la dose oubliée et prenez la dose suivante au moment habituel. Ne prenez pas de dose double pour compenser la dose que vous avez oublié de prendre.
Si vous arrêtez de prendre FÉBUXOSTAT ARROW 80 mg, comprimé pelliculé
N’arrêtez pas de prendre FÉBUXOSTAT ARROW sans l'avis de votre médecin, même si vous vous sentez mieux. Si vous arrêtez de prendre FÉBUXOSTAT ARROW, votre taux d’acide urique sanguin peut augmenter à nouveau et vos symptômes peuvent s’aggraver en raison de la formation de nouveaux cristaux d'urate dans et autour de vos articulations et de vos reins.
Si vous avez d'autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin ou votre pharmacien.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Arrêtez de prendre ce médicament et contactez immédiatement votre médecin ou allez au service des urgences le plus proche si les effets indésirables rares (pouvant affecter jusqu’à 1 personne sur 1 000) suivants se produisent, étant donné qu’ils peuvent être suivis d’une grave réaction allergique :
· réactions anaphylactiques, hypersensibilité au médicament (voir également rubrique 2 « Avertissements et précautions ») ;
· éruptions cutanées pouvant engager le pronostic vital caractérisées par la formation de cloques et une desquamation des muqueuses internes, telles que celles de la bouche et des organes génitaux, ulcères douloureux dans la bouche et/ou des aires génitales, accompagnés de fièvre, maux de gorge et fatigue (syndrome de Stevens-Johnson/nécrolyse épidermique toxique (syndrome de Lyell)), ou par un gonflement des ganglions lymphatiques, une augmentation de la taille du foie, une hépatite (pouvant aller jusqu’à une insuffisance hépatique), une augmentation du nombre de globules blancs dans le sang (syndrome d’hypersensibilité médicamenteuse avec éosinophilie et symptômes systémiques – syndrome DRESS) (voir rubrique 2) ;
· éruptions cutanées généralisées.
Les effets indésirables fréquents (pouvant affecter jusqu’à 1 personne sur 10) sont :
· anomalie du bilan hépatique ;
· diarrhées ;
· maux de tête ;
· éruption (incluant des éruptions de type varié, voir ci-dessous les rubriques « peu fréquent » et « rare ») ;
· nausée ;
· augmentation des symptômes de la goutte ;
· gonflement localisé dû à une rétention de liquide dans les tissus (œdème) ;
· difficulté respiratoire ;
· démangeaison ;
· extrémités douloureuses, douleurs/endolorissement des muscles/articulations ;
· fatigue.
Les autres effets indésirables qui ne sont pas mentionnés ci-dessus sont listés ci-dessous.
Effets indésirables peu fréquents (pouvant affecter jusqu’à 1 personne sur 100) :
· diminution de l’appétit, modification du taux de sucre dans le sang (diabète, dont un symptôme peut être une soif excessive), augmentation du taux de lipides dans le sang, prise de poids ;
· perte du désir sexuel ;
· troubles du sommeil, somnolence ;
· sensation d’engourdissement, sensation de picotements, diminution ou altération des sensations (hypoesthésie, hémiparésie ou paresthésie), altération du sens du goût, diminution de l’odorat (hyposmie) ;
· anomalie de l’électrocardiogramme, battements cardiaques irréguliers ou rapides, perception des battements du cœur (palpitations) ;
· bouffées de chaleur (rougeur du visage ou du cou), augmentation de la pression artérielle, saignement (hémorragie, observé que chez les patients sous chimiothérapie pour des maladies du sang) ;
· toux, gêne ou douleur dans la poitrine, inflammation du canal nasal et/ou de la gorge (infections des voies respiratoires supérieures), bronchite, infection des voies respiratoires inférieures ;
· bouche sèche, maux de ventre/gêne abdominale ou gaz, douleur abdominale haute, remontées acides/indigestion, constipation, défécations plus fréquentes, vomissements, gêne gastrique ;
· éruption cutanée avec démangeaisons, urticaire, inflammation de la peau, dépigmentation de la peau, petits boutons rouges ou violets sur la peau, petits boutons rouges aplatis sur la peau, zones rouges sur la peau recouvertes de petites cloques confluentes, éruption, zones de rougeur et boutons sur la peau, transpiration accrue, sueurs nocturnes, alopécie, rougissement de la peau (érythème), psoriasis, eczéma, autre type d’affection de la peau ;
· crampe musculaire, faiblesse musculaire, bursite ou arthrite (inflammation des articulations habituellement accompagnée de douleurs, d’un gonflement et/ou d’une raideur), douleurs dorsales, spasme musculaire, raideur musculaire et/ou articulaire ;
· sang dans l’urine, émission d’urine anormalement fréquente, anomalie de l’analyse d’urine (augmentation du taux de protéines dans l’urine), réduction de l’aptitude des reins à fonctionner correctement, infection de l’appareil urinaire ;
· douleur à la poitrine, gêne dans la poitrine ;
· calculs dans la vésicule biliaire ou dans les canaux biliaires (cholélithiase) ;
· augmentation du taux d’hormone stimulant la thyroïde (TSH) dans le sang ;
· modifications des paramètres biochimiques du sang ou du nombre de cellules sanguines (anomalies des analyses de sang) ;
· calculs rénaux ;
· difficultés à avoir une érection ;
· diminution de l’activité de la glande thyroïde ;
· vision trouble, changement de la vision,
· nez qui coule ;
· ulcération buccale ;
· inflammation du pancréas : les symptômes communs sont des douleurs abdominales, des nausées et des vomissements ;
· envie impérieuse d’uriner ;
· douleur ;
· malaise ;
· augmentation de l’INR ;
· contusion ;
· gonflement des lèvres.
Effets indésirables rares (pouvant affecter jusqu’à 1 personne sur 1 000) :
· lésions des cellules musculaires, pouvant être graves dans de rares cas. Ces lésions peuvent provoquer des troubles musculaires. Plus particulièrement, si dans le même temps, vous ne vous sentez pas bien ou si vous avez de la température, cela peut être dû à une dégradation musculaire anormale. Contactez immédiatement votre médecin si vous ressentez une douleur, une fragilité ou une faiblesse musculaire ;
· gonflement important des tissus profonds de la peau, touchant particulièrement le contour des yeux, les parties génitales, les mains, les pieds ou la langue, pouvant être associé à une difficulté soudaine à respirer ;
· fièvre élevée accompagnée d’une éruption cutanée de type rougeole, d’un gonflement des ganglions lymphatiques, d’une augmentation de la taille du foie, d’une hépatite (pouvant aller jusqu’à une insuffisance hépatique), d’une augmentation du nombre de globules blancs dans le sang (leucocytose avec ou sans éosinophilie) ;
· éruption de type varié (avec des boutons blancs, des cloques, des cloques contenant du pus, desquamation de la peau, éruption de type rougeole), rougeur de la peau étendue, destruction et décollement bulleux de l’épiderme et des muqueuses, entraînant une exfoliation et une possible infection généralisée (syndrome de Stevens-Johnson/nécrolyse épidermique toxique (syndrome de Lyell)) ;
· nervosité ;
· sensation de soif ;
· perte de poids, augmentation de l’appétit, perte de l’appétit incontrôlée (anorexie) ;
· numération de la formule sanguine anormalement basse (des globules blancs ou des globules rouges ou des plaquettes) ;
· modification ou diminution de la quantité d’urine dues à une inflammation des reins (néphrite tubulo-interstitielle) ;
· inflammation du foie (hépatite) ;
· jaunissement de la peau (jaunisse) ;
· lésion du foie ;
· augmentation du taux de créatine phosphokinases dans le sang (témoin de lésion musculaire) ;
· mort subite cardiaque ;
· diminution du nombre de globules rouges (anémie) ;
· dépression ;
· troubles du sommeil ;
· perte du goût ;
· sensations de brûlures ;
· vertiges ;
· insuffisance circulatoire ;
· infection pulmonaire (pneumonie) ;
· plaies dans la bouche, inflammation de la bouche ;
· perforation gastro-intestinale ;
· syndrome de la coiffe des rotateurs ;
· pseudopolyarthrite rhizomélique ;
· sensations de chaud ;
· perte soudaine de la vision dur à l’occlusion d’une artère dans l’œil.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : www.signalement-sante.gouv.fr
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER FÉBUXOSTAT ARROW 80 mg, comprimé pelliculé ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur l’emballage après EXP. La date de péremption fait référence au dernier jour de ce mois.
Ce médicament ne nécessite pas de précautions particulières de conservation.
Ne jetez aucun médicament au tout à l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient FÉBUXOSTAT ARROW 80 mg, comprimé pelliculé
· La substance active est :
Fébuxostat (sous forme hémihydratée)............................................................................. 80 mg
Pour un comprimé pelliculé
· Les autres composants sont :
Noyau du comprimé : lactose monohydraté, cellulose microcristalline (grade 101), croscarmellose sodique, hydroxypropylcellulose, cellulose microcristalline (grade 102), silice colloïdale anhydre, stéarate de magnésium.
Pelliculage du comprimé : alcool polyvinylique, dioxyde de titane (E171), macrogol 3350, talc, oxyde de fer jaune (E172).
Qu’est-ce que FÉBUXOSTAT ARROW 80 mg, comprimé pelliculé et contenu de l’emballage extérieur
FÉBUXOSTAT ARROW 80 mg, comprimé pelliculé est disponible en boîtes de 14, 28, 30, 42, 56, 84 et 98 comprimés pelliculés.
Toutes les présentations peuvent ne pas être commercialisées.
Titulaire de l’autorisation de mise sur le marché
26 AVENUE TONY GARNIER
69007 LYON
Exploitant de l’autorisation de mise sur le marché
26 AVENUE TONY GARNIER
69007 LYON
RUA JOAO DE DEUS, 19
AMADORA 2700-487
PORTUGAL
OU
ARROW GENERIQUES
26 AVENUE TONY GARNIER
69007 LYON
OU
APL SWIFT SERVICES (MALTA) LIMITED
HF26, HAL FAR INDUSTRIAL ESTATE, HAL FAR
BIRZEBBUGIA, BBG 3000
MALTE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Ce médicament est autorisé dans les Etats membres de l'Espace Economique Européen sous les noms suivants : Conformément à la réglementation en vigueur.
[À compléter ultérieurement par le titulaire]
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
< {MM/AAAA}>< {mois AAAA}.>
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).