VALGANCICLOVIR ARROW
Dosage: 450mg

")
")
. La carte Vitale est une marque déposée par l'assurance maladie")
Cliquez sur un pictogramme pour aller directement à la rubrique le concernant.
Pour plus d'information sur les pictogrammes, consultez l'aide.
Indications thérapeutiques
Classe pharmacothérapeutique - code ATC : J05AB14.
VALGANCICLOVIR ARROW appartient à un groupe de médicaments qui agissent directement pour empêcher la multiplication des virus. La substance active des comprimés, le valganciclovir, est transformée dans l’organisme en ganciclovir. Le ganciclovir empêche la multiplication d’un virus appelé cytomégalovirus (CMV) et l’invasion des cellules saines par celui-ci. Chez les patients dont le système immunitaire est affaibli, le CMV peut provoquer une infection des organes. Celle-ci peut mettre en danger la vie des patients.
VALGANCICLOVIR ARROW est utilisé :
· Pour le traitement des infections à CMV de la rétine de l’œil chez les patients adultes atteints d’un syndrome d’immunodéficience acquise (SIDA). Une infection à CMV de la rétine de l’œil peut provoquer des troubles visuels et même une cécité.
· Pour prévenir les infections à CMV chez les adultes et les enfants qui ne sont pas infectés par le CMV et qui ont reçu une greffe d’organe provenant d’une personne qui était infectée par le CMV.
Groupe(s) générique(s)
Ce médicament appartient au(x) groupe(s) générique(s) suivants :
Composition en substances actives
-
 Comprimé (Composition pour un comprimé)
Comprimé (Composition pour un comprimé)
-
> valganciclovir
 450 mg
450 mg
-
 sous forme de : chlorhydrate de valganciclovir
sous forme de : chlorhydrate de valganciclovir
-
-
> valganciclovir
Présentations
> plaquette(s) polyamide aluminium PVC-Aluminium de 60 comprimé(s)
Code CIP : 34009 300 089 6 6Déclaration de commercialisation : 30/11/2016
Cette présentation est agréée aux collectivités
Taux de remboursement : 65%
Service médical rendu (SMR) 
Ce médicament étant un générique, le SMR n'a pas été évalué par la commission de la transparence (CT), il est possible de se référer à la /aux spécialité(s) de référence du groupe générique auquel appartient ce médicament (cliquez ici pour aller à la rubrique des groupes génériques)
Amélioration du service médical rendu (ASMR)
Ce médicament étant un générique, l'ASMR n'a pas été évalué par la commission de la transparence (CT), il est possible de se référer à la /aux spécialité(s) de référence du groupe générique auquel appartient ce médicament (cliquez ici pour aller à la rubrique des groupes génériques)
Autres informations (cliquer pour afficher)
- Titulaire de l'autorisation : ARROW GENERIQUES
- Conditions de prescription et de délivrance :
- Statut de l'autorisation : Valide
- Type de procédure : Procédure décentralisée
- Code CIS : 6 688 153 2
ANSM - Mis à jour le : 01/06/2023
VALGANCICLOVIR ARROW 450 mg, comprimé pelliculé
2. COMPOSITION QUALITATIVE ET QUANTITATIVE 
Valganciclovir (sous forme de chlorhydrate de valganciclovir)................................................ 450 mg
Pour un comprimé pelliculé.
Excipient à effet notoire : aucun
Pour la liste complète des excipients, voir rubrique 6.1.
Comprimés pelliculés de forme ovale, biconvexes, de couleur rose, portant en relief la mention « H » sur une face et la mention « 96 » sur l'autre face. La taille est de 16,8 mm x 7,9 mm.
4.1. Indications thérapeutiques
VALGANCICLOVIR ARROW est indiqué en traitement prophylactique des infections à CMV chez les patients adultes et les enfants (dès la naissance jusqu’à 18 ans) CMV-négatifs ayant bénéficié d'une transplantation d’organe solide à partir d’un donneur CMV-positif.
4.2. Posologie et mode d'administration
Le valganciclovir est rapidement et largement métabolisé en ganciclovir après administration orale. Un traitement par le valganciclovir per os à 900 mg deux fois par jour équivaut à un traitement par le ganciclovir administré par voie IV à 5 mg/kg deux fois par jour.
Traitement de la rétinite à cytomégalovirus (CMV)
Patients Adultes
Traitement d’attaque de la rétinite à CMV :
Pour les patients présentant une rétinite à CMV active, la posologie recommandée est de 900 mg de valganciclovir (deux comprimés de VALGANCICLOVIR ARROW dosés à 450 mg) deux fois par jour pendant 21 jours ; les comprimés doivent être pris avec des aliments, dans la mesure du possible. Un traitement d’attaque prolongé peut accroître le risque de toxicité médullaire (voir rubrique 4.4).
Traitement d’entretien de la rétinite à CMV :
À la suite d’un traitement d’attaque ou chez les patients présentant une rétinite à CMV non évolutive, la posologie recommandée est de 900 mg de valganciclovir (deux comprimés de VALGANCICLOVIR ARROW dosés à 450 mg) une fois par jour ; les comprimés doivent être pris dans la mesure du possible avec des aliments. Le traitement d’attaque peut être répété chez les patients dont la rétinite s’aggrave. Cependant, la possibilité d’une résistance virale au médicament devra être envisagée.
La durée du traitement d’entretien doit être déterminée au cas par cas.
Population pédiatrique
La tolérance et l’efficacité de valganciclovir dans le traitement de la rétinite à CMV n’ont pas été établies au cours d’études cliniques adéquates et bien contrôlées menées chez des patients pédiatriques.
Traitement prophylactique de la maladie à CMV dans la transplantation d'organes solides
Patients Adultes
Chez les patients ayant reçu une greffe de rein, la posologie recommandée est de 900 mg (deux comprimés de Valganciclovir Arrow dosés à 450 mg) une fois par jour, le traitement doit être initié dans les 10 jours suivant la transplantation et poursuivi jusqu'à 100 jours après celle-ci. La prophylaxie peut être poursuivie jusqu’à 200 jours après la transplantation (voir rubriques 4.4, 4.8 et 5.1).
Chez les patients transplantés d’un organe solide autre que le rein, la posologie recommandée est de 900 mg (deux comprimés de Valganciclovir Arrow dosés à 450 mg) une fois par jour, le traitement devant être initié dans les 10 jours suivant la transplantation et poursuivi jusqu'à 100 jours après celle-ci.
Lorsque cela est possible, les comprimés doivent être administrés pendant un repas.
Population pédiatrique
Chez les patients pédiatriques ayant reçu dès la naissance une greffe d’organe solide, qui présentent un risque de développer une maladie à CMV, la dose quotidienne unique recommandée de valganciclovir est calculée en fonction de la surface corporelle (SC) et de la clairance de la créatinine (ClCr), calculée à partir de la formule de Schwartz (ClCrS), selon l’équation suivante :
Posologie pédiatrique (mg) = 7 x SC x ClCrS (voir la formule de la surface corporelle de Mosteller et la formule de la clairance de la créatinine de Schwartz ci-dessous).
Si la clairance de la créatinine calculée selon la formule de Schwartz dépasse 150 ml/min/1,73 m², alors une valeur maximale de 150 ml/min/1,73 m² doit être utilisée dans l’équation :
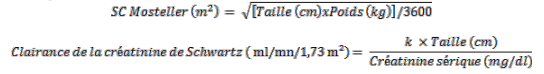
Où k = 0,45* pour les patients < 2 ans ; 0,55 pour les garçons de 2 à < 13 ans et pour les filles de 2 à 16 ans ; et 0,7 pour les garçons de 13 à 16 ans. Pour les patients de plus de 16 ans, se référer à la posologie de l’adulte.
Les valeurs de k sont basées sur la méthode de Jaffé pour la mesure de la créatinine sérique et peuvent nécessiter une correction lorsque des méthodes enzymatiques sont utilisées.
*Pour certaines sous-populations, une diminution de la valeur de k peut également être nécessaire (p.ex., chez les patients pédiatriques présentant un faible poids à la naissance).
Pour les patients pédiatriques ayant reçu une greffe de rein, la dose quotidienne unique recommandée en mg (7 x SC x ClCrS) doit être administrée dans les 10 jours suivant la transplantation et se poursuivre jusqu’à 200 jours après celle-ci.
Pour les patients pédiatriques ayant reçu une greffe d’organe solide autre que le rein, la dose quotidienne unique recommandée en mg (7 x SC x ClCrS) doit être administrée dans les 10 jours suivant la transplantation et se poursuivre jusqu’à 100 jours après celle-ci.
Toutes les doses calculées doivent être arrondies à l’incrément de 25 mg le plus proche pour la dose réelle administrable. Si la dose calculée dépasse 900 mg, il faut administrer une dose maximale de 900 mg. La solution buvable est la formulation recommandée dans la mesure où elle permet d’administrer une dose calculée selon la formule ci-dessus. Cependant, les comprimés pelliculés de valganciclovir peuvent être utilisés si la dose calculée est comprise dans un intervalle de 10 % des dosages disponibles en comprimés, et si le patient est en mesure d’avaler des comprimés. Par exemple, si la dose calculée est comprise entre 405 mg et 495 mg, un comprimé à 450 mg peut être administré.
Il est recommandé de contrôler les taux de créatinine sérique régulièrement et de prendre en compte les variations de taille et de poids corporel, puis d’adapter la posologie en conséquence au cours de la période de prophylaxie.
Instructions posologiques particulières
Population pédiatrique
La posologie administrée aux patients pédiatriques ayant reçu une greffe d’organe solide est déterminée à partir de la fonction rénale du patient, et de sa surface corporelle.
Patients âgés :
La sécurité et l’efficacité n’ont pas été établies dans cette population. Aucune étude n’a été menée chez les patients âgés de plus de 65 ans. Étant donné que la fonction rénale diminue avec l’âge, le valganciclovir doit être administré aux patients âgés avec une attention particulière sur l’état de leur fonction rénale (voir le tableau ci-dessous) (voir rubrique 5.2).
Insuffisants rénaux :
La créatinine sérique ou la clairance de la créatinine estimée doivent être étroitement surveillées. La posologie doit être ajustée en fonction de la clairance de créatinine comme indiqué dans le tableau ci-dessous (voir rubriques 4.4 et 5.2).
La clairance de la créatinine (ml/min) peut être estimée à partir de la valeur de créatinine sérique au moyen des formules suivantes :
(140 - âge [ans]) x (poids corporel [kg])
Chez l’homme = -----------------------------------------------------------
(72) x (0,011 x créatinine sérique [micromoles/l])
Chez la femme = 0,85 x valeur chez l’homme.
|
ClCr (ml/min) |
Posologie du valganciclovir Traitement d'attaque |
Posologie du valganciclovir Traitement d'entretien - Prophylaxie |
|
≥ 60 |
900 mg (2 comprimés) 2 fois par jour |
900 mg (2 comprimés) 1 fois par jour |
|
40 - 59 |
450 mg (1 comprimé) 2 fois par jour |
450 mg (1 comprimé) 1 fois par jour |
|
25 - 39 |
450 mg (1 comprimé) 1 fois par jour |
450 mg (1 comprimé) tous les 2 jours |
|
10 - 24 |
450 mg (1 comprimé) tous les 2 jours |
450 mg (1 comprimé) 2 fois par semaine |
|
< 10 |
Non recommandé |
Non recommandé |
Patients hémodialysés :
Pour les patients sous hémodialyse (ClCr < 10 ml/min), il n’est pas possible de recommander une posologie. Par conséquent, le valganciclovir comprimé pelliculé ne doit pas être utilisé chez ces patients (voir rubriques 4.4 et 5.2).
Insuffisants hépatiques :
La sécurité et l’efficacité de comprimés de valganciclovir n’ont pas été établies chez l’insuffisant hépatique (voir rubrique 5.2).
Patients présentant une leucopénie, neutropénie, anémie, thrombopénie ou pancytopénie sévère :
Voir rubrique 4.4 avant de débuter le traitement.
En cas de dégradation significative de la numération formule sanguine au cours du traitement par le valganciclovir, l’administration de facteurs de croissance hématopoïétique et/ou l’interruption du traitement doivent être envisagées (voir rubrique 4.4).
Mode d’administration
Le valganciclovir est administré par voie orale et doit, dans la mesure du possible, être pris avec des aliments (voir rubrique 5.2).
Pour les patients pédiatriques qui ne sont pas en mesure d’avaler les comprimés pelliculés, une autre forme pharmaceutique peut être utilisée, par exemple, la poudre pour solution buvable.
Précautions à prendre avant de manipuler ou d’administrer le médicament
Les comprimés ne doivent être ni cassés ni broyés. Le valganciclovir étant considéré comme potentiellement tératogène et carcinogène chez l’homme, des précautions doivent être prises pour la manipulation des comprimés cassés (voir rubrique 4.4). Éviter le contact direct des comprimés cassés ou broyés avec la peau ou les muqueuses. En cas de contact, laver abondamment au savon et à l’eau ; rincer abondamment les yeux à l’eau stérile ou à l’eau courante en l’absence d’eau stérile.
VALGANCICLOVIR ARROW est contre-indiqué durant l’allaitement (voir rubrique 4.6).
4.4. Mises en garde spéciales et précautions d'emploi
En raison de la similarité de la structure chimique du ganciclovir et de celle de l’aciclovir et du penciclovir, une réaction d’hypersensibilité croisée entre ces médicaments est possible. VALGANCICLOVIR ARROW doit donc être prescrit avec prudence chez les patients présentant une hypersensibilité connue à l’aciclovir ou au penciclovir (ou à leurs prodrogues respectives le valaciclovir ou le famciclovir).
Mutagénicité, tératogénicité, carcinogénicité, fertilité et contraception
Avant l’initiation du traitement par le valganciclovir, les patients doivent être avertis des risques potentiels pour le fœtus. Les études chez l’animal ont montré que le ganciclovir est mutagène, tératogène, aspermatogénique et carcinogène et altère la fertilité chez les femelles. Le valganciclovir doit donc être considéré comme potentiellement tératogène et carcinogène chez l’homme, et peut causer des malformations congénitales et des cancers (voir rubrique 5.3). D’après des études cliniques et non cliniques, il est également probable que le valganciclovir provoque une inhibition temporaire ou définitive de la spermatogenèse. Les femmes en âge de procréer doivent être informées de la nécessité d’utiliser une méthode de contraception efficace durant le traitement et pendant au moins 30 jours après le traitement. Les hommes doivent être informés de la nécessité d’avoir recours à une méthode de contraception mécanique durant le traitement, et pendant au moins 90 jours après le traitement, sauf s’il est certain que la partenaire n'est pas à risque de procréer (voir rubriques 4.6, 4.8 et 5.3).
Le valganciclovir est potentiellement carcinogène et toxique à long terme sur la reproduction.
Myélosuppression
Une leucopénie, une neutropénie, une anémie, une thrombopénie, une pancytopénie, une insuffisance médullaire et une anémie d'origine centrale, toutes de caractère sévère, ont été observées chez des patients traités par valganciclovir (et ganciclovir). Aucun traitement ne doit être initié si le nombre absolu des polynucléaires neutrophiles est inférieur à 500 cellules/µl, ou le nombre des plaquettes est inférieur à 25 000/µl, ou le taux d’hémoglobine est inférieur à 8 g/dl (voir rubriques 4.2 et 4.8).
Lorsque la prophylaxie est prolongée au-delà de 100 jours, le risque éventuel de développer une leucopénie et une neutropénie doit être pris en compte (voir sections 4.2, 4.8 et 5.1).
Le valganciclovir doit être utilisé avec précaution chez les patients ayant une cytopénie hématologique préexistante ou des antécédents de cytopénie hématologique d’origine médicamenteuse, ainsi que chez les patients sous radiothérapie.
Il est recommandé de surveiller régulièrement la numération formule sanguine et le nombre des plaquettes durant le traitement. Une surveillance hématologique plus étroite peut être nécessaire chez les insuffisants rénaux et chez les enfants, au minimum à chaque fois que le patient se rend à un rendez-vous dans l’établissement dans lequel il a été greffé. Chez les patients développant une leucopénie, une neutropénie, une anémie et/ou une thrombopénie de caractère sévère, il est recommandé d’envisager un traitement par facteurs de croissance hématopoïétique et/ou une interruption du traitement (voir rubrique 4.2).
Différence de biodisponibilité avec ganciclovir par voie orale
La biodisponibilité du ganciclovir après administration unique de 900 mg de valganciclovir est d’environ 60 % comparée à environ 6 % après administration orale de 1 000 mg de ganciclovir (en gélules). Une exposition excessive au ganciclovir peut entraîner des effets indésirables pouvant menacer le pronostic vital. Il est donc conseillé de respecter scrupuleusement les posologies recommandées lors de l’instauration du traitement, lors du passage du traitement d’attaque au traitement d’entretien, ainsi que chez les patients qui passent du ganciclovir oral au valganciclovir, étant donné que ce dernier ne peut être substitué au ganciclovir en gélules sur la base d’un comprimé pour une gélule. Les patients passant des gélules de ganciclovir au valganciclovir doivent être avertis du risque de surdosage s’ils prennent un nombre de comprimés de valganciclovir supérieur au nombre prescrit (voir rubriques 4.2 et 4.9).
Insuffisance rénale
Chez les insuffisants rénaux, la posologie doit être ajustée en fonction de la clairance de la créatinine (voir rubriques 4.2 et 5.2).
Le valganciclovir en comprimé pelliculé ne doit pas être utilisé chez les patients sous hémodialyse (voir rubriques 4.2 et 5.2).
Utilisation avec d’autres médicaments
Des convulsions ont été rapportées chez des patients recevant de l’imipénème-cilastatine et du ganciclovir. Le valganciclovir ne doit pas être utilisé en même temps que l’imipénème-cilastatine, sauf si les bénéfices attendus justifient les risques encourus (voir rubrique 4.5).
Chez les patients traités par le valganciclovir et par (a) la didanosine, (b) des médicaments connus comme myélosuppresseurs (par exemple, la zidovudine), ou (c) des substances modifiant la fonction rénale, la recherche de signes de toxicité accrue devra être étroitement surveillée (voir rubrique 4.5).
L’essai clinique contrôlé utilisant le valganciclovir dans le traitement prophylactique de la maladie à CMV en cas de transplantation tel que décrit dans la rubrique 5.1 ne comprenait pas de patients ayant bénéficié d’une greffe de poumon ou d’intestin. Par conséquent, l’expérience chez ces patients transplantés reste limitée.
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Interactions médicamenteuses avec le valganciclovir
Aucune étude in vivo d'interactions médicamenteuses avec le valganciclovir n'a été réalisée. Le valganciclovir étant rapidement et largement métabolisé en ganciclovir, les interactions médicamenteuses observées avec le ganciclovir sont attendues avec le valganciclovir.
Interactions médicamenteuses avec le ganciclovir
Interactions pharmacocinétiques
Probénécide
L'administration de probénécide et de ganciclovir per os a entraîné une diminution statistiquement significative de la clairance rénale du ganciclovir (20 %), provoquant une augmentation statistiquement significative de l'exposition au ganciclovir (40 %). Ces modifications étaient compatibles avec un mécanisme d'interaction faisant intervenir une compétition pour la sécrétion tubulaire rénale. Par conséquent, les patients recevant du probénécide et du valganciclovir doivent être étroitement surveillés en ce qui concerne la toxicité du ganciclovir.
Didanosine
Les concentrations plasmatiques de didanosine ont systématiquement augmenté en cas d’administration concomitante de ganciclovir intraveineux. À des doses intraveineuses de 5 et 10 mg/kg/jour, une augmentation de l’ASC de la didanosine allant de 38 % à 67 % a été observée, confirmant une interaction pharmacocinétique durant l’administration concomitante de ces médicaments. Aucun effet significatif sur les concentrations du ganciclovir n’a été constaté. Les patients doivent être étroitement surveillés en ce qui concerne les signes liés à la toxicité de la didanosine comme la pancréatite (voir rubrique 4.4).
Autres antirétroviraux
Les isoenzymes du cytochrome P450 ne jouent aucun rôle dans la pharmacocinétique du ganciclovir. En conséquence, les interactions pharmacocinétiques avec les inhibiteurs de la protéase et les inhibiteurs non nucléosidiques de la transcriptase inverse ne sont pas attendues.
Le gangiclovir étant excrété par le rein via la filtration glomérulaire et la sécrétion tubulaire active (rubrique 5.2), la co-administration du valganciclovir avec des antiviraux qui ont la même voie d’élimination par sécrétion tubulaire peut modifier les concentrations plasmatiques en valganciclovir et/ou en médicaments co-administrés. Certains exemples incluent des analogues nucléos(t)idiques inhibiteurs de la transcriptase inverse (NRTIs) (comprenant ceux utilisés pour le traitement du VHB), p.ex. la lamivudine, l’emtricitabine, le ténofovir, l’adéfovir et l’entécavir. La clairance rénale du ganciclovir peut aussi être inhibée par nephrotoxicité induite des médicaments tels que : cidofovir, foscarnet, NRTI (p.ex. le ténofovir et l’adefovir). L’utilisation concomitante du valganciclovir avec l’un de ces médicaments doit être envisagée uniquement si les bénéfices attendus justifient les risques encourus (voir la rubrique 4.4)
Interactions pharmacodynamiques
Imipénème-cilastatine
Des convulsions ont été rapportées chez des patients recevant de façon concomitante du ganciclovir et de l'imipénème-cilastatine. Ces médicaments ne doivent pas être utilisés de façon concomitante, sauf si les bénéfices attendus justifient les risques encourus (voir rubrique 4.4).
Zidovudine
La zidovudine et le ganciclovir ont tous les deux la capacité d’induire une neutropénie et une anémie. Une interaction pharmacodynamique peut survenir durant l’administration concomitante de ces médicaments. Certains patients ne pourront pas tolérer une thérapie concomitante à pleine dose (voir rubrique 4.4)
Interactions médicamenteuses potentielles
La toxicité peut être augmentée si le ganciclovir/valganciclovir est administré de façon concomitante avec d’autres médicaments dont l’effet myélosuppresseur est connu ou qui sont associés à une altération de la fonction rénale. Cela inclut les analogues nucléosidiques (dont zidovudine, didanosine, stavudine), ainsi que les analogues nucléotidiques (dont ténofovir et adéfovir), les immunosuppresseurs (dont ciclosporine, tacrolimus, mycophénolate mofétil), les agents antinéoplasiques (dont doxorubicine, vinblastine, vincristine, hydroxyurée) et les agents antiinfectieux (triméthoprime/sulfamide, dapsone, amphotéricine B, flucytosine, pentamidine). Par conséquent, ces médicaments doivent seulement être considérés pour une utilisation concomitante avec le valganciclovir si les bénéfices attendus dépassent les risques encourus (voir rubrique 4.4).
4.6. Fertilité, grossesse et allaitement
Contraception chez les hommes et les femmes
En raison du potentiel de toxicité pour la reproduction et de tératogénicité, les femmes en âge de procréer doivent être informées de la nécessité d’utiliser une méthode de contraception efficace pendant le traitement et pendant au moins 30 jours après le traitement. Les hommes, quant à eux, doivent être informés de la nécessité d’avoir recours à une méthode de contraception mécanique pendant le traitement, et pendant au moins 90 jours suivant le traitement par valganciclovir, sauf s’ils sont certains que leur partenaire ne présente aucun risque de grossesse (voir rubriques 4.4 et 5.3).
Grossesse
La sécurité d’emploi du valganciclovir chez la femme enceinte n’a pas été établie. Son métabolite actif, le ganciclovir, traverse facilement le placenta humain. Compte tenu de son mécanisme d’action pharmacologique et des effets nocifs sur la reproduction observés avec le ganciclovir dans les études chez l’animal (voir rubrique 5.3), il existe un risque théorique de tératogénicité chez l’homme.
Le valganciclovir ne doit pas être utilisé pendant la grossesse, sauf si les bénéfices attendus pour la mère justifient les risques tératogènes potentiels pour le fœtus.
Chez l’être humain, aucune donnée n'est disponible sur la sécrétion de ganciclovir dans le lait maternel ; toutefois, l’éventualité d’une excrétion du ganciclovir dans le lait maternel, provoquant des réactions indésirables graves chez le nourrisson, ne peut être écartée.
Des données chez l’animal indiquent que le ganciclovir est excrété dans le lait de rates allaitantes. Par conséquent, l’allaitement maternel doit être arrêté pendant le traitement par valganciclovir (voir rubriques 4.3 et 5.3).
Fertilité
Une étude clinique de faible envergure chez des patients transplantés rénaux recevant du valganciclovir dans le cadre d’une prophylaxie des infections à CMV jusqu’à 200 jours a démontré un impact du valganciclovir sur la spermatogenèse avec une diminution de la densité et de la motilité des spermatozoïdes mesurés à la fin du traitement. Cet effet semble être réversible, et environ 6 mois après l’arrêt du valganciclovir, la densité moyenne et la motilité des spermatozoïdes retrouvent des niveaux comparables à ceux observés chez les témoins non traités.
Lors d’études pratiquées chez l’animal, le ganciclovir a altéré la fertilité chez les souris mâles et femelles et il a été montré qu’il inhibait la spermatogenèse et induisait une atrophie testiculaire chez la souris, le rat et le chien à des doses considérées comme cliniquement appropriées.
Sur la base d’études cliniques et non cliniques, il est probable que le ganciclovir (et le valganciclovir) puisse provoquer une inhibition temporaire ou permanente de la spermatogenèse chez l’homme (voir rubriques 4.4 et 5.3).
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Les effets sur l’aptitude à conduire des véhicules et à utiliser des machines n’ont pas été étudiés.
Des convulsions, des étourdissements, et une confusion ont été rapportés avec le traitement par valganciclovir et/ou ganciclovir. S’ils surviennent, ces effets peuvent affecter les tâches nécessitant une capacité de vigilance, notamment l’aptitude du patient à conduire des véhicules ou à utiliser des machines.
a. Résumé du profil de sécurité
Le valganciclovir est une prodrogue du ganciclovir, qui est rapidement et largement métabolisée en ganciclovir après administration orale. Lors d’un traitement par le valganciclovir, la survenue des effets indésirables imputables à l’utilisation du ganciclovir peut être attendue. Tous les effets indésirables observés dans les études cliniques du valganciclovir avaient déjà été observés avec le ganciclovir. Par conséquent, les effets indésirables rapportés avec le ganciclovir intraveineux ou oral (la formulation par voie orale n’est plus disponible) ou avec le valganciclovir sont inclus dans le tableau des effets indésirables ci-dessous.
Chez les patients traités par le valganciclovir/ganciclovir, les effets indésirables les plus graves et les plus fréquents sont hématologiques, notamment neutropénie, anémie et thrombopénie (voir rubrique 4.4).
Les fréquences présentées dans le tableau des effets indésirables proviennent d’une population de patients (n=1704) recevant un traitement d’entretien par du ganciclovir ou du valganciclovir. A l’exception de la réaction anaphylactique, l’agranulocytose et la granulocytopénie, dont les fréquences sont tirées de l’expérience post-AMM. Les effets indésirables sont présentés par MedDRA systeme organ classe (SOC). Les catégories de fréquence sont définies en utilisant la convention suivante : très fréquent (≥ 1/10), fréquent (≥ 1 /100 à < 1/10), peu fréquent (≥ 1 /1000 à < 1 /100), rare (≥ 1/10 000 à < 1/1 000), très rare (< 1/10 000).
Le profil de sécurité du ganciclovir/valganciclovir est cohérent entre les populations de patients infectés par le VIH et les patients transplantés à l’exception du décollement de la rétine qui a uniquement été rapporté chez les patients VIH avec une rétinite à CMV. Cependant, il y a des différences dans la fréquence de certaines réactions. Le valganciclovir est associé à un risque plus élevé de diarrhée comparé au ganciclovir intraveineux. La fièvre, les infections à candida, la dépression, les neutropénies sévères (NAN <500/µL) et les réactions cutanées sont plus fréquemment rapportées chez les patients infectés par le VIH. Des dysfonctionnements rénaux et hépatiques sont rapportés plus fréquemment chez les receveurs de greffe d’organe.
b. Tableau des effets indésirables
|
Effets indésirables (MedDRA) Système Organ Classe (SOC) |
Catégorie de fréquence |
|
|
Infections et infestations : |
||
|
Candidoses, y compris buccales |
Très fréquent |
|
|
Infection des voies respiratoires supérieures |
||
|
Septicémie |
Fréquent |
|
|
Grippe |
||
|
Infection des voies urinaires |
||
|
Cellulite |
||
|
Affections hématologiques et du système lymphatique : |
||
|
Neutropénie |
Très fréquent |
|
|
Anémie |
||
|
Thrombocytopénie |
Fréquent |
|
|
Leucopénie |
||
|
Pancytopénie |
||
|
Insuffisance médullaire |
Peu fréquent |
|
|
Anémie aplasique |
Rare |
|
|
Agranulocytose* |
||
|
Granulocytopénie* |
||
|
Affections du système immunitaire : |
||
|
Hypersensibilité |
Fréquent |
|
|
Réaction anaphylactique* |
Rare |
|
|
Troubles du métabolisme et de la nutrition : |
||
|
Diminution de l’appétit |
Très fréquent |
|
|
Diminution du poids |
Fréquent |
|
|
Affections psychiatriques : |
||
|
Dépression |
Fréquent |
|
|
État confusionnel |
||
|
Anxiété |
||
|
Agitation |
Peu fréquent |
|
|
Troubles psychotiques |
||
|
Troubles de la pensée |
||
|
Hallucinations |
||
|
Affections du système nerveux : |
||
|
Céphalées |
Très fréquent |
|
|
Insomnie |
Fréquent |
|
|
Neuropathie périphérique |
||
|
Sensations vertigineuses |
||
|
Paresthésies |
||
|
Hypoesthésie |
||
|
Convulsion |
||
|
Dysgueusie (perturbation du goût) |
||
|
Tremblements |
Peu fréquent |
|
|
Affections oculaires : |
||
|
Altération de la vision |
Fréquent |
|
|
Décollement de la rétine** |
||
|
Corps flottants du vitré |
||
|
Douleur oculaire |
||
|
Conjonctivite |
||
|
Œdème maculaire |
||
|
Affections de l’oreille et du labyrinthe : |
||
|
Douleur auriculaire |
Fréquent |
|
|
Surdité |
Peu fréquent |
|
|
Affections cardiaques : |
||
|
Peu fréquent |
|
|
Affections vasculaires : |
||
|
Hypotension |
Fréquent |
|
|
Affections respiratoires, thoraciques et médiastinales : |
||
|
Toux |
Très fréquent |
|
|
Dyspnée |
||
|
Affections gastro-intestinales : |
||
|
Diarrhée |
Très fréquent |
|
|
Nausées |
||
|
Vomissements |
||
|
Douleurs abdominales |
||
|
Dyspepsie |
Fréquent |
|
|
Flatulences |
||
|
Douleurs abdominales hautes |
||
|
Constipation |
||
|
Ulcération buccale |
||
|
Dysphagie |
||
|
Distension abdominale |
||
|
Pancréatite |
||
|
Affections hépatobiliaires : |
||
|
Augmentation de la phosphatase alcaline sérique |
Fréquent |
|
|
Anomalie de la fonction hépatique |
||
|
Augmentation de l’aspartate aminotransférase |
||
|
Augmentation de l’alanine aminotransférase |
||
|
Affections de la peau et du tissu sous-cutané : |
||
|
Dermatite |
Très fréquent |
|
|
Sueurs nocturnes |
Fréquent |
|
|
Prurit |
||
|
Rash |
||
|
Alopécie |
||
|
Peau sèche |
Peu fréquent |
|
|
Urticaire |
||
|
Affections musculo-squelettiques et systémiques : |
||
|
Dorsalgies |
Fréquent |
|
|
Myalgies |
||
|
Arthralgies |
||
|
Spasmes musculaires |
||
|
Affections du rein et des voies urinaires : |
||
|
Altération de la fonction rénale |
Fréquent |
|
|
Diminution de la clairance de la créatinine |
||
|
Augmentation de la créatininémie |
||
|
Insuffisance rénale |
Peu fréquent |
|
|
Hématurie |
||
|
Affections des organes de reproduction et du sein : |
||
|
Infertilité masculine |
Peu fréquent |
|
|
Troubles généraux et anomalies au site d’administration : |
||
|
Pyrexie |
Très fréquent |
|
|
Fatigue |
||
|
Douleur |
Fréquent |
|
|
Frissons |
||
|
Malaise |
||
|
Asthénie |
||
|
Douleur thoracique |
Peu fréquent |
|
* Les fréquences de ces effets indésirables sont tirées de données post-AMM
** Le détachement de la rétine a uniquement été rapporté chez les patients VIH traités pour une rétinite à CMV
Description de certains effets indésirables
Neutropénie
Le risque de neutropénie ne peut être anticipé sur la base du nombre de polynucléaires neutrophiles avant traitement. La neutropénie apparaît habituellement durant la première ou la deuxième semaine du traitement d’induction. La numération des polynucléaires neutrophiles se normalise habituellement en 2 à 5 jours après l’arrêt du médicament ou une réduction de sa dose (voir rubrique 4.4).
Thrombopénie
Le risque de survenue d’une thrombopénie est plus élevé chez les patients dont la numération plaquettaire est initialement basse (< 100 000/µl). Les patients présentant une immunosuppression iatrogène due à un traitement par des médicaments immunosuppresseurs sont exposés à un risque plus élevé de thrombopénie que les patients atteints du sida (voir rubrique 4.4). Une thrombopénie sévère peut être associée à une hémorragie pouvant engager le pronostic vital.
Influence de la durée du traitement ou de l’indication sur les effets indésirables
Des cas de neutropénie sévère (NAN <500/µL) sont plus fréquemment observés chez les patients atteints de rétinite à CMV (14%) sous traitement par valganciclovir, par ganciclovir par voie orale ou intraveineuse que chez les patients ayant reçu une greffe d’organe solide sous traitement par valganciclovir ou ganciclovir oral. Chez les patients recevant du valganciclovir ou du ganciclovir oral jusqu’au 100ème jour après la transplantation, l’incidence de la neutropénie sévère était respectivement de 5% et de 3%, alors que chez les patients recevant du valganciclovir jusqu’au 200ème jour après la transplantation, l’incidence de la neutropénie sévère était de 10%.
En comparaison avec les patients atteints de rétinite à CMV, une augmentation plus importante de la créatinine sérique a été observée chez les patients ayant reçu une greffe d’organe solide traités jusqu’au 100ème jour ou 200ème jour après la transplantation, à la fois par valganciclovir et par ganciclovir oral. Cependant, l’insuffisance rénale est une caractéristique fréquente chez le patient ayant reçu une greffe d’organe solide.
Le profil global de sécurité d’emploi du valganciclovir est resté similaire lorsque la prophylaxie a été prolongée jusqu’à 200 jours chez des patients à haut risque ayant reçu une greffe de rein. Des cas de leucopénie ont été rapportés selon une incidence légèrement supérieure dans le bras 200 jours, tandis que les incidences de la neutropénie, de l’anémie et de la thrombocytopénie ont été similaires dans les deux bras.
c. Population pédiatrique
Le valganciclovir a été étudié chez 179 patients pédiatriques (âgés de 3 semaines à 16 ans) ayant reçu une greffe d’organe plein et qui présentaient un risque de développer une maladie à CMV et chez 133 nouveau-nés souffrant d’une maladie congénitale à CMV symptomatique (âgés de 2 à 31 jours), la durée d’exposition au ganciclovir allant de 2 à 200 jours.
Les effets indésirables les plus fréquemment rapportés au cours du traitement administré pendant les essais cliniques pédiatriques ont été les suivants : diarrhée, nausées, neutropénie, leucopénie et anémie
Pour les patients ayant reçu une greffe d’organe solide, le profil global de sécurité d’emploi chez les patients pédiatriques était identique à celui des adultes. Des cas de neutropénie ont, également, été signalés avec une incidence légèrement supérieure dans les deux études réalisées chez des patients pédiatriques ayant reçu une greffe d’organe plein par rapport aux adultes. Toutefois, aucune corrélation entre la neutropénie et les événements indésirables infectieux n’a été établie au sein de la population pédiatrique. Un risque plus élevé de cytopénies chez les nouveau-nés et les nourrissons justifie une surveillance plus étroite de la numération globulaire au sein de ces groupes d’âge (voir section 4.4).
Chez les patients pédiatriques ayant bénéficié d’une greffe de rein, l’exposition prolongée au valganciclovir jusqu’à 200 jours n’a pas été associée à une augmentation globale de l’incidence des événements indésirables. L’incidence des neutropénies sévères (PNN < 500/µl) a été plus élevée chez les patients pédiatriques ayant bénéficié d’une greffe de rein et traités pendant 200 jours que chez les patients pédiatriques traités pendant 100 jours, ainsi que chez les patients adultes ayant reçu une greffe de rein traités pendant 100 ou 200 jours (voir rubrique 4.4).
Seules des données limitées sont disponibles chez les nouveau-nés ou les nourrissons présentant des infections congénitales à CMV symptomatiques et traités par valganciclovir. Cependant, le profil de sécurité d’emploi semble être conforme au profil de sécurité d’emploi connu du valganciclovir/ganciclovir.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet :https://signalement.social-sante.gouv.fr/.
Expérience du surdosage avec le valganciclovir et le ganciclovir intraveineux
On peut s’attendre à ce qu’un surdosage en valganciclovir puisse se traduire par une toxicité rénale accrue (voir rubriques 4.2 et 4.4).
Des cas de surdosage de ganciclovir intraveineux, dont certains d’issue fatale, ont été rapportés durant les études cliniques et après mise sur le marché. Dans certains cas, aucun événement indésirable n’a été rapporté. La majorité des patients a présenté un ou plusieurs des événements indésirables suivants :
· Toxicité hématologique : myélosuppression incluant pancytopénie, insuffisance médullaire, leucopénie, neutropénie, granulopénie.
· Hépatotoxicité : hépatite, troubles de la fonction hépatique.
· Néphrotoxicité : aggravation de l’hématurie chez un patient avec insuffisance rénale préexistante, insuffisance rénale aiguë, élévation de la créatinine.
· Toxicité gastro-intestinale : douleur abdominale, diarrhée, vomissements.
· Neurotoxicité : tremblements généralisés, convulsions.
L’hémodialyse et l’hydratation pourraient être bénéfiques en permettant une réduction des concentrations plasmatiques chez les patients ayant reçu un surdosage en valganciclovir (voir rubrique 5.2).
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Mécanisme d’action
Le valganciclovir est un ester de L-valyle (prodrogue) du ganciclovir. Après administration orale, le valganciclovir est rapidement et largement métabolisé en ganciclovir par des estérases intestinales et hépatiques. Le ganciclovir est un analogue de synthèse de la 2’-désoxyguanosine, qui inhibe la réplication des virus du groupe herpès in vitro et in vivo. Les virus humains sensibles comprennent le cytomégalovirus humain (CMV), les Herpesvirus simplex 1 et 2 (HSV-1 et HSV-2), les Herpesvirus humains 6, 7 et 8 (HHV-6, HHV-7, HHV-8), le virus d’Epstein-Barr (EBV), le virus varicelle-zona (VZV) et le virus de l’hépatite B (VHB).
Dans les cellules infectées par le CMV, le ganciclovir est initialement phosphorylé en ganciclovir monophosphate par la protéine kinase virale pUL97. Une phosphorylation ultérieure par des kinases cellulaires conduit au ganciclovir triphosphate, qui subit ensuite un métabolisme intracellulaire lent. Le ganciclovir triphosphate a été mis en évidence dans des cellules infectées par HSV et CMV, avec des demi-vies respectives de 18 heures et 6 à 24 heures, après élimination du ganciclovir extracellulaire. Étant donné que la phosphorylation est largement dépendante de la kinase virale, la phosphorylation du ganciclovir se produit préférentiellement dans les cellules infectées par le virus.
L’activité virustatique du ganciclovir est due à l’inhibition de la synthèse de l’ADN viral par : (a) inhibition compétitive de l’incorporation de désoxyguanosine-triphosphate dans l’ADN par l’ADN polymérase virale, et (b) incorporation de ganciclovir triphosphate dans l’ADN viral provoquant l’arrêt de ou une forte limitation de l’allongement de l’ADN viral.
Activité antivirale
L’activité antivirale in vitro, mesurée par la CI50 du ganciclovir contre le CMV, est comprise entre 0,08 μM (0,02 μg/ml) et 14 μM (3,5 μg/ml).
L’effet antiviral clinique du valganciclovir a été démontré dans le traitement de patients atteints du SIDA et présentant une rétinite à CMV nouvellement diagnostiquée. L’excrétion urinaire de CMV a été diminuée de 46 % des patients (32/69) à l’entrée dans l’étude à 7 % des patients (4/55) après quatre semaines de traitement par valganciclovir.
Efficacité clinique et sécurité
Patients adultes
Traitement de la rétinite à CMV :
Des patients présentant une rétinite à CMV nouvellement diagnostiquée ont été randomisés dans une étude pour recevoir un traitement d’attaque par le valganciclovir (900 mg deux fois par jour) ou par le ganciclovir intraveineux (5 mg/kg deux fois par jour). La proportion des patients avec progression photographique de la rétinite à CMV à la 4ème semaine était comparable dans les deux groupes traités, une progression étant constatée chez 7 patients sur 70 dans le groupe ganciclovir intraveineux et chez 7 patients sur 71 dans le groupe valganciclovir.
Après le traitement d’attaque, tous les patients de cette étude ont reçu un traitement d’entretien par le valganciclovir à la dose de 900 mg une fois par jour. Le délai moyen (médian) entre la randomisation et la progression de la rétinite à CMV a été de 226 (160) jours dans le groupe ayant reçu un traitement d’attaque et un traitement d’entretien par le valganciclovir et de 219 (125) jours dans le groupe ayant reçu un traitement d’attaque par le ganciclovir intraveineux et un traitement d’entretien par le valganciclovir.
Prophylaxie de la maladie à CMV dans des transplantations d'organes :
Une étude clinique randomisée en double aveugle avec comparateur actif a été conduite chez des patients ayant reçu une greffe de cœur, de foie ou de rein (les patients ayant reçu une greffe de poumon ou d’intestin n’ont pas été inclus dans cette étude), à haut risque de maladie à CMV (D+/R-) et qui ont reçu du valganciclovir (900 mg une fois par jour) ou du ganciclovir oral (1 000 mg trois fois par jour), le traitement étant débuté dans les 10 jours suivant la transplantation et poursuivi jusqu'à 100 jours après celle-ci. L'incidence de la maladie à CMV (syndrome à CMV + invasion tissulaire) au cours des six premiers mois suivant la transplantation était de 12,1 % dans le bras valganciclovir (n = 239) contre 15,2 % dans le bras ganciclovir oral (n = 125).
La grande majorité des cas est survenue après l'arrêt de la prophylaxie (100 jours après transplantation) avec une apparition des cas en moyenne plus tardive dans le bras valganciclovir que dans le bras ganciclovir oral. L'incidence du rejet aigu au cours des six premiers mois était de 29,7 % chez les patients randomisés dans le bras valganciclovir contre 36,0 % dans le bras ganciclovir oral avec une incidence équivalente de perte du greffon se produisant chez 0,8 % des patients dans chacun des bras.
Une étude clinique contrôlée versus placebo, en double aveugle, a été conduite chez 326 patients ayant reçu une greffe de rein à haut risque de maladie à CMV (D+/R-) pour évaluer l’efficacité et la sécurité de la prolongation de la durée de la prophylaxie de 100 jours à 200 jours après la transplantation. Les patients ont été randomisés (1:1) pour recevoir les comprimés de valganciclovir (900 mg une fois par jour) dans les 10 jours suivant la transplantation et soit jusqu’à 200 jours après celle-ci, soit jusqu’à 100 jours après celle-ci suivi de 100 jours de placebo.
La proportion de patients qui ont développé une maladie à CMV dans les 12 premiers mois après la transplantation est présentée dans le tableau ci-dessous.
Pourcentage de patients ayant reçu une greffe de rein et ayant développé une maladie à CMV1, population en ITT dans les 12 mois A
|
|
Valganciclovir 900 mg une fois par jour 100 jours (N = 163) |
Valganciclovir 900 mg une fois par jour 200 jours (N = 155) |
Différence entre les groupes de traitement |
|
Maladie à CMV confirmée ou suspectée2 |
71 (43,6 %) [35,8 % ; 51,5 %] |
36 (23,2 %) [16,8 % ; 30,7 %] |
20,3 % [9,9 % ; 30,8 %] |
|
Maladie à CMV confirmée |
60 (36,8 %) [29,4 % ; 44,7 %] |
25 (16,1 %) [10,7 % ; 22,9 %] |
20,7 % [10,9 % ; 30,4 %] |
1 La maladie à CMV est définie par un syndrome à CMV ou par une maladie à CMV avec invasion tissulaire. 2 Une maladie à CMV confirmée est une maladie à CMV prouvée cliniquement. Une maladie à CMV a été considérée pour les patients sans évaluation à 52 semaines et pour ceux sans confirmation de maladie à CMV avant cette période.
A Les résultats à 24 mois étaient comparables à ceux obtenus à 12 mois : maladie à CMV confirmée ou suspectée chez 48,5 % des patients dans le bras de prophylaxie à 100 jours contre 34,2 % des patients dans le bras de la prophylaxie à 200 jours, la différence entre les groupes de traitement était de 14,3 % [3,2 % ; 25,3 %].
Le nombre de transplantés rénaux à haut risque de maladie à CMV qui ont développé une maladie à CMV après une prophylaxie par valganciclovir de 200 jours après transplantation est significativement moins élevé comparé au nombre de patients ayant reçu une prophylaxie par valganciclovir de 100 jours après la transplantation.
Le taux de survie du greffon ainsi que l’incidence d’un rejet aigu prouvé par biopsie est similaire dans les deux groupes de traitement. Le taux de survie du greffon à 12 mois après transplantation était de 98,2 % (160/163) pour une durée de prophylaxie de 100 jours et 98,1 % (152/155) pour une durée de prophylaxie de 200 jours. Quatre cas supplémentaires de perte de greffon ont été rapportés, tous dans le groupe de traitement à 100 jours, jusqu’à 24 mois après la transplantation. L’incidence d’un rejet aigu prouvé par biopsie à 12 mois après transplantation était de 17,2 % (28/163) pour une durée de prophylaxie de 100 jours et de 11,0 % (17/155) pour une durée de prophylaxie de 200 jours. Un cas supplémentaire a été rapporté jusqu’à 24 mois après la transplantation dans le groupe de traitement à 200 jours.
Résistance virale
Des virus résistants au ganciclovir peuvent apparaître après traitement chronique par le valganciclovir, par sélection de mutations au niveau du gène de la kinase virale (UL97) responsable de la monophosphorylation du ganciclovir et/ou au niveau du gène de la polymérase virale (UL54). Dans des isolats cliniques, les sept substitutions canoniques UL97 (M460V/I, H520Q, C592G, A594V, L595S, C603W) sont les substitutions les plus fréquemment associées à une résistance.
Les virus contenant des mutations au niveau du gène UL97 sont résistants au ganciclovir seul, tandis que les virus contenant des mutations au niveau du gène UL54 sont résistants au ganciclovir, mais peuvent présenter une résistance croisée à d’autres antiviraux qui ciblent également la polymérase virale.
Traitement de la rétinite à CMV :
Une analyse génotypique des CMV dans des isolats de polynucléaires (PMNL) obtenus chez 148 patients ayant une rétinite à CMV inclus dans une étude clinique a révélé des taux de présence de mutations UL97 de 2,2 %, 6,5 %, 12,8 % et 15,3 % après respectivement 3, 6, 12 et 18 mois de traitement par le valganciclovir.
Prophylaxie de la maladie à CMV en transplantation d'organes :
Étude de comparateur actif
La résistance a été étudiée par une analyse génotypique des CMV dans des PMNL collectés i) à J100 (fin de la prophylaxie) et ii) en cas de suspicion de maladie à CMV jusqu'à 6 mois après transplantation. Parmi les 245 patients randomisés dans le bras ganciclovir, 198 échantillons à J100 ont été recueillis pour être testés et aucune mutation de résistance au ganciclovir n'a été observée. Ceci est à comparer aux deux mutations de résistance au ganciclovir détectées dans les 103 échantillons testés (1,9 %) des patients du bras comparateur (ganciclovir oral).
Parmi les 245 patients randomisés dans le bras valganciclovir, 50 échantillons issus de patients pour lesquels une maladie à CMV était suspectée ont été testés et aucune mutation de résistance n'a été observée. Parmi les 127 patients randomisés dans le bras comparateur ganciclovir, 29 échantillons issus de patients pour lesquels une maladie à CMV était suspectée ont été testés et deux mutations de résistance ont été observées, soit une incidence de résistance de 6,9 %.
Étude de prolongation de la prophylaxie de 100 jours à 200 jours après transplantation
Une analyse génotypique a été conduite sur les gènes viraux UL54 et UL97 extraits de 72 patients qui ont répondu au critère d’analyse de la résistance : patients ayant une charge virale positive (>600 copies/ml) à la fin de la prophylaxie et/ou patients ayant une maladie à CMV confirmée jusqu’à 12 mois (52 semaines) après la transplantation. Trois patients dans chaque groupe de traitement avaient une mutation de résistance au ganciclovir connue.
Population pédiatrique
Traitement de la rétinite à CMV :
L’Agence européenne des médicaments a accordé une dérogation à l’obligation de réaliser des études cliniques sur le valganciclovir dans tous les sous-groupes de la population pédiatrique dans le traitement de l’infection à CMV chez des patients immunodéprimés (rubrique 4.2 pour les informations concernant l’usage pédiatrique).
Prévention de la maladie à CMV en transplantation d’organes :
Dans une étude de pharmacocinétique et de sécurité de phase II chez des enfants ayant bénéficié d'une transplantation d'organe solide (âgés de 4 mois à 16 ans, n = 63), le valganciclovir a été administré une fois par jour jusqu'à 100 jours selon un algorithme de posologie pédiatrique (voir rubrique 4.2) permettant d’obtenir des expositions similaires à celles de l’adulte (voir rubrique 5.2). Le suivi après le traitement était de 12 semaines. Le statut sérologique CMV D/R initial était D+/R- dans 40 % des cas, D+/R+ dans 38 %, D-/R+ dans 19 % et D-/R- dans 3 % des cas. La présence du virus CMV a été rapportée chez 7 patients. Les effets indésirables observés étaient de nature similaire à ceux rapportés chez l’adulte (voir rubrique 4.8). Une étude de phase IV sur la tolérance menée chez des enfants ayant bénéficié d’une greffe de rein (âgés de 1 à 16 ans, n = 57) recevant du valganciclovir une fois par jour jusqu’à 200 jours, selon l’algorithme posologique (voir rubrique 4.2), a mis en évidence une faible incidence du CMV. Le suivi après le traitement était de 24 semaines. Le statut sérologique CMV D/R initial était de D+/R+ dans 45 % des cas, D+/R- dans 39 %, D-/R+ dans 7 %, D-/R- dans 7 % et ND/R+ dans 2 % des cas. Une virémie à CMV a été rapportée chez trois patients et un syndrome à CMV a été suspecté chez un patient, mais non confirmé par une PCR effectuée sur le CMV par le laboratoire central. Les effets indésirables observés étaient de nature similaire à ceux rapportés chez l’adulte (voir rubrique 4.8).
Ces données soutiennent l’extrapolation des données d’efficacité des adultes par rapport aux enfants, et permettent de fournir des recommandations posologiques pour les patients pédiatriques.
Une étude de pharmacocinétique de phase I sur la tolérance menée chez des patients ayant bénéficié d’une transplantation cardiaque (âgés de 3 semaines à 125 jours, n = 14), qui ont reçu une dose quotidienne unique de valganciclovir selon l’algorithme posologique pédiatrique (voir rubrique 4.2) pendant deux jours consécutifs, a mis en évidence des expositions similaires à celles constatées chez l’adulte (voir rubrique 5.2). Le suivi après le traitement était de 7 jours. Le profil d’innocuité était conforme à celui observé au cours d’autres études menées chez des enfants et des adultes, bien que le nombre de patients et l’exposition au valganciclovir aient été limités au cours de cette étude.
Infection congénitale à CMV
L’efficacité et la tolérance du ganciclovir et/ou du valganciclovir ont été étudiées dans deux études réalisées chez des nouveau-nés et des nourrissons présentant une infection congénitale à CMV symptomatique.
Au cours de la première étude, la pharmacocinétique et la sécurité d’une dose unique de valganciclovir (plage de dose 14-16-20 mg/kg/dose) ont été étudiées chez 24 nouveau-nés (âgés de 8 à 34 jours) atteints d’une infection congénitale à CMV symptomatique (voir rubrique 5.2). Les nouveau-nés ont reçu 6 semaines de traitement antiviral. Parmi les 24 patients, 19 ont reçu jusqu’à 4 semaines de traitement par valganciclovir oral suivi de ganciclovir par voie I.V au cours des 2 semaines suivantes. Les 5 autres patients ont reçu le ganciclovir par voie I.V. pendant la majeure partie de la période d’étude.
Lors de la seconde étude, l’efficacité et la tolérance du traitement par valganciclovir de six semaines par rapport à six mois ont été étudiées chez 109 nourrissons, âgés de 2 à 30 jours, présentant une maladie congénitale à CMV symptomatique. Tous les nourrissons ont reçu du valganciclovir par voie orale à la posologie de 16 mg/kg, deux fois par jour, pendant six semaines. Après six semaines de traitement, les nourrissons ont été randomisés selon un rapport de 1/1 pour poursuivre le traitement par valganciclovir à la même posologie ou recevoir un placebo afin de compléter les six mois de traitement.
Le valganciclovir n’est actuellement pas recommandé dans cette indication thérapeutique. Le schéma des études et les résultats obtenus sont trop limités pour conclure sur l’efficacité et la sécurité du valganciclovir.
5.2. Propriétés pharmacocinétiques
La proportionnalité de la dose par rapport à l’ASC du ganciclovir après administration de dose de valganciclovir allant de 450 à 2625 mg n’a été démontrée que chez le sujet s’étant alimenté.
Absorption
Le valganciclovir est une prodrogue du ganciclovir. Il est bien absorbé à partir du tube digestif et rapidement et largement métabolisé en ganciclovir dans la paroi intestinale et le foie. L’exposition systémique au valganciclovir est transitoire et faible. La biodisponibilité du ganciclovir dérivé du valganciclovir administré par voie orale est d’environ 60 % pour l’ensemble des populations de patients étudiées et l’exposition résultante au ganciclovir est similaire à celle obtenue après administration intraveineuse de ganciclovir (voir ci-dessous). En comparaison, la biodisponibilité du ganciclovir après une administration orale de 1 000 mg (en gélules) est de 6 à 8 %.
Valganciclovir chez les patients VIH positif, CMV positif :
L’exposition systémique de patients VIH positif, CMV positif après administration deux fois par jour de ganciclovir et de valganciclovir pendant une semaine est la suivante :
|
Paramètre |
Ganciclovir (5 mg/kg, IV) n = 18 |
Valganciclovir (900 mg, per os) n = 25 |
|
|
Ganciclovir |
Valganciclovir |
||
|
ASC(0-12 h) (µg.h/ml) |
28,6 ± 9,0 |
32,8 ± 10,1 |
0,37 ± 0,22 |
|
Cmax (μg/ml) |
10,4 ± 4,9 |
6,7 ± 2,1 |
0,18 ± 0,06 |
Il a été démontré que l’efficacité du ganciclovir pour accroître le délai avant progression de la rétinite à CMV est corrélée à l’exposition systémique (ASC).
Valganciclovir chez les patients ayant subi une transplantation d'organe solide :
L’exposition systémique à l'équilibre de patients ayant subi une transplantation d’organe solide après administration quotidienne orale de ganciclovir et de valganciclovir est la suivante :
|
Paramètre |
Ganciclovir (1 000 mg 3x/j) n = 82 |
Valganciclovir (900 mg, une fois par jour) n = 161 |
|
Ganciclovir |
||
|
ASC(0-24 h) (µg.h/ml) |
28,0 ± 10,9 |
46,3 ± 15,2 |
|
Cmax (μg/ml) |
1,4 ± 0,5 |
5,3 ± 1,5 |
Conformément à l'algorithme de réduction de la posologie en fonction de l'état de la fonction rénale, l'exposition systémique au ganciclovir était similaire chez les patients ayant reçu une greffe de cœur, de rein ou de foie après administration orale de valganciclovir.
Effet de l’alimentation :
Lorsque le valganciclovir était administré avec des aliments à la dose recommandée de 900 mg, des augmentations ont été constatées à la fois pour l’ASC moyenne du ganciclovir (environ 30 %) et pour la Cmax moyenne du ganciclovir (environ 14 %), par comparaison à l’administration à jeun. En outre, les variations interindividuelles de l’exposition au ganciclovir diminuent lorsque le valganciclovir est pris avec des aliments. Le valganciclovir a toujours été administré avec des aliments au cours des études cliniques. Par conséquent, il est recommandé d’administrer le valganciclovir avec des aliments (voir rubrique 4.2).
Distribution
En raison de la conversion rapide du valganciclovir en ganciclovir, la liaison du valganciclovir aux protéines n’a pas été déterminée. Le volume de distribution (Vd) du ganciclovir à l’équilibre après administration intraveineuse était de 0,680 ± 0,161 l/kg (n = 114). Pour le ganciclovir intraveineux, le volume de distribution est corrélé au poids corporel avec des valeurs du volume de distribution à l’état d’équilibre allant de 0,54 à 0,87 L/kg. Le ganciclovir pénètre dans le liquide céphalorachidien. La liaison aux protéines plasmatiques était 1% à 2% pour des concentrations de ganciclovir de 0,5 et 51 µg/mL.
Biotransformation
Le valganciclovir est rapidement et largement métabolisé en ganciclovir ; aucun autre métabolite n’a été détecté. Le ganciclovir est peu métabolisé.
Élimination
Après administration orale de valganciclovir, le médicament est rapidement hydrolysé en ganciclovir. Le ganciclovir est éliminé de la circulation systémique par filtration glomérulaire et sécrétion tubulaire active La demi-vie du ganciclovir issu du valganciclovir est de 4,1 ± 0,9 heures chez des patients séropositifs pour le VIH et le CMV. Chez les patients dont la fonction rénale est normale, plus de 90 % de la dose de ganciclovir administrée par voie intraveineuse était retrouvée sous forme inchangée dans l’urine au cours des 24 heures suivant l’administration. Chez les patients dont la fonction rénale est normale, la concentration plasmatique maximale de ganciclovir après administration de valganciclovir décroit avec une demi-vie allant de 0,4 h à 2,0 h.
Pharmacocinétique dans des situations cliniques particulières
Population pédiatrique
Dans une étude de pharmacocinétique et de sécurité de phase II chez des enfants ayant bénéficié d'une transplantation d'organe solide (âgés de 4 mois à 16 ans, n = 63), le valganciclovir a été administré une fois par jour jusqu'à 100 jours. Les paramètres pharmacocinétiques obtenus sont similaires quel que soit l’organe transplanté et l’âge et sont comparables à ceux obtenus chez l’adulte. Un modèle pharmacocinétique de population a suggéré que la biodisponibilité était d'environ 60 %. La clairance est positivement corrélée à la fois à la surface corporelle et à la fonction rénale
Lors d’une étude de pharmacocinétique de phase I sur la tolérance menée chez des enfants ayant bénéficié d’une greffe de cœur (âgés de 3 mois à 125 jours, n = 14), le valganciclovir a été administré une fois par jour pendant deux jours d’étude. Les propriétés pharmacocinétiques de population ont permis d’estimer une biodisponibilité moyenne de 64 %.
Une comparaison entre les résultats de ces deux études et les résultats pharmacocinétiques obtenus dans la population adulte a montré que les intervalles de l’ASC0 à 24 h sont très semblables pour toutes les tranches d’âge, y compris les adultes. Les valeurs moyennes de l’ASC0 à 24 h et de la Cmax sont également similaires pour toutes les tranches d’âge pédiatriques < 12 ans, bien qu’il ait été observé une tendance à la diminution des valeurs moyennes de l’ASC0 à 24 h et de la Cmax dans l’ensemble de la population pédiatrique, montrant une corrélation avec l’augmentation de l’âge. Cette tendance a été plus prononcée pour les valeurs moyennes de la clairance et de la demi-vie (t½). Cependant, ces résultats étaient attendus dans la mesure où la clairance est influencée par les variations du poids, de la taille et de la fonction rénale associés à la croissance des patients, comme cela est indiqué par la modélisation des propriétés pharmacocinétiques de population.
Le tableau suivant récapitule les intervalles des ASC0 à 24 h estimés par le modèle pour le ganciclovir à partir de ces deux études, ainsi que les valeurs moyennes et l’écart type de l’ASC0-24 h, de la Cmax, de la clairance et de la t½ pour les groupes d’âge pédiatriques pertinents par rapport aux valeurs obtenues chez les adultes.
|
Paramètres PK |
Adultes* |
Enfants |
|||
|
|
≥ 18 ans (n=160) |
< 4 mois (n = 14) |
4 mois - ≤ 2 ans (n=17) |
> 2 - < 12 ans (n=21) |
≥ 12 ans – 16 ans (n=25) |
|
ASC0-24h (μg.h/ml) |
46.3 ± 15.2 |
68.1±19.8 |
64.3±29.2 |
59.2 ±15.1 |
50.3 ± 15.0 |
|
Intervalle d’ASC0-24h |
15.4 - 116.1 |
34 - 124 |
34 - 152 |
36-108 |
22-93 |
|
Cmax (μg/ml) |
5.3 ± 1.5 |
10.5 ±3.36 |
10.3 ± 3.3 |
9.4 ±2.7 |
8.0 ±2.4 |
|
Clairance (l/h) |
12.7 ± 4.5 |
1.25 ± 0.473 |
2.5 ± 2.4 |
4.5 ± 2.9 |
6.4 ±2.9 |
|
t1/2 (h) |
6.5 ± 1.4 |
1.97± 0.185 |
3.1 ± 1.4 |
4.1 ±1.3 |
5.5 ±1.1 |
* Extrait du rapport d'étude PV 16000
La dose unique quotidienne de valganciclovir dans les deux études décrites ci-dessus a été déterminée sur la base de la surface corporelle (SC) et de la clairance de la créatinine (ClCr) dérivée de la formule de Schwartz modifiée et a été calculée en utilisant l’équation ci-dessous :
Les propriétés pharmacocinétiques du ganciclovir, après l’administration du valganciclovir, ont également été évaluées au cours de deux études menées chez des nouveau-nés et des nourrissons présentant une maladie congénitale à CMV symptomatique. Lors de la première étude, 24 nouveau-nés âgés de 8 à 34 jours ont reçu 6 mg/kg de ganciclovir intraveineux deux fois par jour. Les patients ont ensuite été traités par valganciclovir oral, la dose de valganciclovir poudre pour solution buvable étant comprise entre 14 mg/kg et 20 mg/kg deux fois par jour pendant 6 semaines. Une dose de 16 mg/kg deux fois par jour de valganciclovir poudre pour solution buvable a produit une exposition au ganciclovir comparable à 6 mg/kg de ganciclovir intraveineux deux fois par jour chez des nouveau-nés et a également permis d’atteindre une exposition au ganciclovir similaire à une dose intraveineuse efficace de 5 mg/kg chez l'adulte.
Lors de la deuxième étude, 109 nouveau-nés, âgés de 2 à 30 jours, ont reçu 16 mg/kg de valganciclovir en poudre pour solution buvable deux fois par jour, pendant six semaines, puis 96 des 109 patients ont été randomisés pour poursuivre le traitement par valganciclovir ou par placebo pendant six mois. Cependant, les valeurs moyennes de l’ASC0 à 12 h ont été inférieures par rapport à celles de la première étude.
Le tableau suivant présente les valeurs moyennes avec les écarts types d'ASC, Cmax et t ½ comparativement aux données obtenues chez l’adulte :
|
Paramètre PK |
Adultes |
Enfants (nouveau-nés et nourrissons) |
||
|
5 mg/kg GAN Dose unique (n=8) |
6 mg/kg GAN Deux fois par jour (n=19) |
16 mg/kg VAL Deux fois par jour (n=19) |
16 mg/kg VAL Deux fois par jour (n=100) |
|
|
ASC0-∞ (µg.h/ml) |
25,4 ± 4,32 |
- |
- |
- |
|
ASC12h (µg.h/ml) |
- |
38,25 ± 42,7 |
30,1 ± 15,1 |
20.85±5.40 |
|
Cmax (μg/ml) |
9,03 ± 1,26 |
12,9 ± 21,5 |
5,44 ± 4,04 |
- |
|
t1/2 (h) |
3,32 ± 0,47 |
2,52 ± 0,55 |
2,98 ± 1,26 |
2.98 ± 1.12 |
GAN = Ganciclovir, i.v.
VAL = Valganciclovir, oral
Ces données sont trop limitées pour conclure en matière d'efficacité ou établir des recommandations posologiques pour les enfants souffrant d'infection congénitale à CMV.
Patients âgés
Aucune étude sur la pharmacocinétique du valganciclovir ou du ganciclovir n’a été menée chez des adultes âgés de plus de 65 ans (voir rubrique 4.2).
Insuffisants rénaux
La pharmacocinétique du ganciclovir a été évaluée après administration d’une dose orale unique de 900 mg de valganciclovir chez 24 individus en bonne santé ayant une insuffisance rénale.
Paramètres pharmacocinétiques du ganciclovir issus de l’administration d’une dose orale unique d’un comprimé de 900 mg de valganciclovir chez des patients ayant un degré d’insuffisance rénale variable :
|
Clairance estimée de la créatinine (mL/min) |
N |
Clairance apparente moyenne (mL/min) ± écart type |
Dernière ASC moyenne (μg∙h/mL) ± écart type |
Demi-vie moyenne (heures) ± écart type |
|
51-70 |
6 |
249 ± 99 |
49,5 ± 22,4 |
4,85 ± 1,4 |
|
21-50 |
6 |
136 ± 64 |
91,9 ± 43,9 |
10,2 ± 4,4 |
|
11-20 |
6 |
45 ± 11 |
223 ± 46 |
21,8 ± 5,2 |
|
£10 |
6 |
12,8 ± 8 |
366 ± 66 |
67,5 ± 34 |
L’altération de la fonction rénale a conduit à une diminution de la clairance du ganciclovir issu du valganciclovir et à une augmentation correspondante de la demi-vie terminale. Par conséquent, une adaptation de la posologie est nécessaire chez l’insuffisant rénal (voir rubriques 4.2 et 4.4).
Patients hémodialysés
Chez les patients hémodialysés, il est impossible de recommander une posologie pour valganciclovir 450 mg, comprimés pelliculés. En effet, la dose individuelle de valganciclovir nécessaire pour ces patients est inférieure au dosage du comprimé (450 mg). De ce fait, valganciclovir 450 mg, comprimés pelliculés ne doit pas être utilisé chez ces patients (voir rubriques 4.2 et 4.4).
Transplantés hépatiques stables
La pharmacocinétique du ganciclovir issu du valganciclovir a été étudiée chez des patients stables ayant reçu une greffe de foie, dans une étude ouverte croisée en quatre segments (n=28). La biodisponibilité du ganciclovir issu du valganciclovir, après administration d’une dose unique de 900 mg de valganciclovir chez des patients ayant pris un repas était approximativement de 60%. L’ASC0-24h était comparable aux valeurs observées avec 5 mg/kg de ganciclovir intraveineux chez des patients ayant reçus une greffe du foie.
Insuffisants hépatiques
La sécurité et l’efficacité de valganciclovir 450 mg, comprimés pelliculés chez l’insuffisant hépatique n’ont pas été étudiées. L’insuffisance hépatique ne devrait pas affecter la pharmacocinétique du ganciclovir puisqu’il est excrété par voie rénale ; de ce fait, aucune recommandation posologique spécifique ne peut être donnée.
Patients atteints de mucoviscidose
Dans une étude de pharmacocinétique de phase I chez des patients ayant bénéficié d’une greffe de poumon avec ou sans mucoviscidose (FK), 31 patients (16FK, 15-nonFK) ont reçu après la transplantation un traitement en prophylaxie par valganciclovir à la posologie de 900 mg/jour. L’étude a démontré que la mucoviscidose n’a pas d’influence significative sur l’exposition systémique au ganciclovir chez les patients ayant bénéficié d’une greffe de poumon. L’exposition au ganciclovir chez ces patients était comparable à celle démontrée comme étant efficace dans la prévention des infections à CMV chez les autres transplantés d’organes solides.
5.3. Données de sécurité préclinique
Le valganciclovir est une prodrogue du ganciclovir et, par conséquent, les effets observés avec le ganciclovir sont également applicables au valganciclovir. La toxicité du valganciclovir dans les études de tolérance précliniques était la même que celle observée avec le ganciclovir et a été induite à des niveaux d’exposition au ganciclovir comparables ou inférieurs à ceux observés chez l’Homme ayant reçu la dose d’induction.
Ces résultats ont montré une toxicité au niveau des gonades (perte des cellules testiculaires) et une néphrotoxicité (urémie, dégénérescence cellulaire) qui étaient irréversibles : une myélotoxicité (anémie, neutropénie, lymphocytopénie) et une toxicité gastro-intestinale (nécrose des cellules de la muqueuse), qui elles, étaient réversibles.
Le ganciclovir a été mutagène dans des cellules de lymphome murin et clastogènes dans des cellules de mammifères. Ces résultats concordent avec la carcinogénicité observée chez la souris avec le ganciclovir. Le ganciclovir est un carcinogène potentiel.
D’autres études ont montré que le ganciclovir est tératogène, embryotoxique, inhibe la spermatogenèse (c’est-à-dire qu’il nuit à la fertilité masculine) et supprime la fertilité féminine.
Des données chez l’animal indiquent que le ganciclovir est excrété dans le lait de rates allaitantes.
Cellulose, microcristalline (grade 101 et grade 102), crospovidone (type B), povidone (K - 30), stéarate de magnésium
Pelliculage
Hypromellose (3 cP, 6 cP), dioxyde de titane (E171), macrogol 400, polysorbate 80, oxyde de fer rouge (E172)
3 ans.
6.4. Précautions particulières de conservation
Ce médicament ne nécessite pas de conditions particulières de conservation.
6.5. Nature et contenu de l'emballage extérieur
Les comprimés pelliculés Valganciclovir Arrow sont disponibles en plaquettes thermoformées de polyamide/ feuille d’aluminium/ PVC-aluminium et en flacons en HDPE avec bouchon en polypropylène et filtre en coton.
Tailles d'emballage :
Plaquettes thermoformées : 10, 30, 60 et 100 comprimés pelliculés.
Flacons HDPE : 60 et 1000 comprimés pelliculés.
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
26 AVENUE TONY GARNIER
69007 LYON
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 301 578 9 3 : plaquettes (polyamide/Aluminium/PVC-Aluminium) de 10 comprimés
· 34009 301 579 0 9 : plaquettes (polyamide/Aluminium/PVC-Aluminium) de 30 comprimés
· 34009 300 089 6 6 : plaquettes (polyamide/Aluminium/PVC-Aluminium) de 60 comprimé(s)
· 34009 300 089 8 0 : plaquettes (polyamide/Aluminium/PVC-Aluminium) de 100 comprimé(s)
· 34009 300 089 9 7 : flacon(s) polyéthylène haute densité (PEHD) de 60 comprimé(s)
· 34009 550 037 3 1 : flacon(s) polyéthylène haute densité (PEHD) de 1000 comprimé(s)
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I.
ANSM - Mis à jour le : 01/06/2023
VALGANCICLOVIR ARROW 450 mg, comprimé pelliculé
Chlorhydrate de valganciclovir
Veuillez lire attentivement cette notice avant de prendre ce médicament car elle contient des informations importantes pour vous.
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin, votre pharmacien ou votre infirmier/ère.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que VALGANCICLOVIR ARROW 450 mg, comprimé pelliculé et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant de prendre VALGANCICLOVIR ARROW 450 mg, comprimé pelliculé ?
3. Comment prendre VALGANCICLOVIR ARROW 450 mg, comprimé pelliculé ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver VALGANCICLOVIR ARROW 450 mg, comprimé pelliculé ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE VALGANCICLOVIR ARROW 450 mg, comprimé pelliculé ET DANS QUELS CAS EST-IL UTILISE ?
Classe pharmacothérapeutique - code ATC : J05AB14.
VALGANCICLOVIR ARROW appartient à un groupe de médicaments qui agissent directement pour empêcher la multiplication des virus. La substance active des comprimés, le valganciclovir, est transformée dans l’organisme en ganciclovir. Le ganciclovir empêche la multiplication d’un virus appelé cytomégalovirus (CMV) et l’invasion des cellules saines par celui-ci. Chez les patients dont le système immunitaire est affaibli, le CMV peut provoquer une infection des organes. Celle-ci peut mettre en danger la vie des patients.
VALGANCICLOVIR ARROW est utilisé :
· Pour le traitement des infections à CMV de la rétine de l’œil chez les patients adultes atteints d’un syndrome d’immunodéficience acquise (SIDA). Une infection à CMV de la rétine de l’œil peut provoquer des troubles visuels et même une cécité.
· Pour prévenir les infections à CMV chez les adultes et les enfants qui ne sont pas infectés par le CMV et qui ont reçu une greffe d’organe provenant d’une personne qui était infectée par le CMV.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT DE PRENDRE VALGANCICLOVIR ARROW 450 mg, comprimé pelliculé ?
Ne prenez jamais VALGANCICLOVIR ARROW 450 mg, comprimé pelliculé :
· si vous êtes allergique au valganciclovir, ganciclovir, ou à l’un des autres composants contenus dans ce médicament, mentionnés dans la rubrique 6.
· si vous allaitez.
Avertissements et précautions
Adressez-vous à votre médecin, pharmacien ou votre infirmier/ère avant de prendre VALGANCICLOVIR ARROW 450 mg, comprimé pelliculé.
· si vous êtes allergique à l’aciclovir, au penciclovir, au valaciclovir ou au famciclovir. Ceux-ci sont d’autres médicaments utilisés afin de traiter les infections virales ;
· si vous avez dans votre sang un nombre faible de globules blancs, de globules rouges ou de plaquettes (petites cellules intervenant dans la coagulation sanguine). Votre médecin effectuera des examens sanguins avant que vous ne commenciez à prendre les comprimés de VALGANCICLOVIR ARROW, et des examens supplémentaires pendant que vous les prendrez ;
· si vous êtes traité par radiothérapie ou si vous êtes hémodialysé ;
· si vous avez des troubles rénaux. Votre médecin peut devoir vous prescrire une dose plus faible et des examens sanguins réguliers pendant le traitement ;
· si vous prenez actuellement des gélules de ganciclovir et si votre médecin souhaite les remplacer par des comprimés de VALGANCICLOVIR ARROW. Il est important que vous ne preniez pas plus de comprimés que le nombre prescrit par votre médecin, car vous risqueriez un surdosage.
Autres médicaments et VALGANCICLOVIR ARROW 450 mg, comprimé pelliculé
Informez votre médecin ou pharmacien si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament, y compris les médicaments délivrés sans ordonnance.
D'autres médicaments pris en même temps que VALGANCICLOVIR ARROW pourraient affecter la quantité de substance active passant dans le sang ou provoquer des effets nocifs. Prévenez votre médecin si vous prenez des médicaments contenant l'une des substances suivantes :
· imipénème-cilastatine (un antibiotique), dont la prise en même temps que VALGANCICLOVIR ARROW peut provoquer des convulsions ;
· zidovudine, didanosine, lamivudine, stavudine, tenofovir, abacavir, emtricitabine ou autres médicaments similaires utilisés pour traiter le SIDA ;
· adéfovir et autres médicaments utilisés pour traiter l’hépatite B
· probénécide (un médicament contre la goutte), dont la prise en même temps que VALGANCICLOVIR ARROW pourrait augmenter la quantité de ganciclovir dans votre sang ;
· mycophénolate mofétil, ciclosporine ou tacrolimus (utilisé après une greffe d'organe) ;
· vincristine, vinblastine, doxorubicine, hydroxyurée ou médicaments similaires utilisés pour traiter le cancer ;
· triméthoprime, association triméthoprime/sulfamide et dapsone (antibiotiques) ;
· pentamidine (médicament utilisé pour traiter les infections parasitaires ou les infections pulmonaires);
· flucytosine ou amphotéricine B (agents antifongiques).
VALGANCICLOVIR ARROW 450 mg, comprimé pelliculé avec des aliments et boissons
Grossesse et allaitement
Vous ne devez pas prendre VALGANCICLOVIR ARROW si vous êtes enceinte sauf si votre médecin vous le prescrit.
Si vous êtes enceinte ou que vous allaitez, si vous pensez être enceinte ou planifiez une grossesse, demandez conseil à votre médecin ou pharmacien avant de prendre ce médicament. La prise de VALGANCICLOVIR ARROW pendant votre grossesse peut être nocive pour l’enfant à naître.
Vous ne devez pas prendre VALGANCICLOVIR ARROW si vous allaitez. Si votre médecin souhaite que vous commenciez un traitement par VALGANCICLOVIR ARROW, vous devez cesser d’allaiter avant de commencer à prendre les comprimés.
Les femmes en âge de procréer doivent utiliser une méthode de contraception efficace pendant toute la durée du traitement par VALGANCICLOVIR ARROW et pendant au moins 30 jours après la fin de leur traitement.
Les hommes dont les partenaires sont en âge de procréer doivent utiliser des préservatifs pendant toute la durée de leur traitement par VALGANCICLOVIR ARROW et doivent continuer à utiliser des préservatifs pendant les 90 jours suivant l’arrêt du traitement.
Demandez conseil à votre médecin ou à votre pharmacien avant de prendre tout médicament.
Conduite de véhicules et utilisation de machines
Il est déconseillé de conduire ou d’utiliser certains outils ou machines si vous avez des étourdissements, des tremblements ou si vous vous sentez fatigué ou confus après la prise de ce médicament.
Demandez conseil à votre médecin ou à votre pharmacien avant de prendre tout médicament.
VALGANCICLOVIR ARROW 450 mg, comprimé pelliculé ne contient pas d’excipient à effet notoire.
3. COMMENT PRENDRE VALGANCICLOVIR ARROW 450 mg, comprimé pelliculé ?
Les comprimés de VALGANCICLOVIR ARROW doivent être manipulés avec précaution. Ils ne doivent être ni cassés ni broyés. Avalez-les entiers et dans la mesure du possible avec des aliments. Si vous avez touché accidentellement des comprimés endommagés, lavez-vous les mains abondamment au savon et à l'eau. Si de la poudre provenant des comprimés entre en contact avec vos yeux, rincez-les avec de l'eau stérile ou à défaut avec de l'eau du robinet.
Vous devez strictement vous conformer au nombre de comprimés prescrits par votre médecin pour éviter un surdosage.
VALGANCICLOVIR ARROW comprimé doit, si possible, être pris au cours d’un repas – voir rubrique 2.
Prévention de la maladie à CMV chez les patients transplantés
Vous devez commencer à prendre le traitement dans les 10 jours suivant la greffe. La posologie habituelle est de deux comprimés pris UNE FOIS par jour. Vous devez continuer le traitement avec cette dose jusqu'à 100 jours après la greffe. Si vous avez reçu une greffe de rein, votre médecin peut vous demander de prendre ce traitement pendant 200 jours.
Traitement de la rétinite à CMV évolutive chez les patients atteints de SIDA (traitement d’attaque)
La posologie habituelle de VALGANCICLOVIR ARROW est de deux comprimés pris DEUX FOIS par jour pendant 21 jours (trois semaines). Ne prenez pas cette dose plus de 21 jours sauf si votre médecin vous l’a indiqué, car cela pourrait augmenter le risque d’effets indésirables éventuels.
Traitement préventif à long terme de l’inflammation évolutive récurrente chez les patients atteints de SIDA présentant une rétinite à CMV (traitement d’entretien)
La posologie habituelle est de deux comprimés pris une fois par jour. Vous devez essayer de prendre les comprimés chaque jour à la même heure. Votre médecin vous dira pendant combien de temps vous devez prendre VALGANCICLOVIR ARROW. Si votre rétinite s’aggrave à cette dose, votre médecin peut vous demander de recommencer un traitement d’attaque (comme ci-dessus) ou peut décider de vous prescrire un traitement différent pour traiter l’infection à CMV.
Personnes âgées
Il n’y a pas d’études concernant l’utilisation de VALGANCICLOVIR ARROW chez les personnes âgées.
Patients avec des troubles rénaux
Si vos reins fonctionnent mal, votre médecin peut vous indiquer de prendre moins de comprimés chaque jour ou de ne les prendre que certains jours chaque semaine. Il est très important que vous respectiez la dose prescrite par votre médecin.
Patients avec des troubles hépatiques
Il n’y a pas d’études concernant l’utilisation de VALGANCICLOVIR ARROW chez les patients avec des troubles hépatiques.
Utilisation chez les enfants et les adolescents :
Prévention de la maladie à CMV chez les patients transplantés
Les enfants doivent commencer à prendre le traitement dans les 10 jours suivant la greffe. La dose prescrite varie en fonction de leur taille et doit être prise UNE FOIS par jour. Votre médecin vous indiquera la dose la plus appropriée en fonction de la taille, du poids et du fonctionnement rénal de votre enfant. L'enfant devra poursuivre cette dose jusqu'à 100 jours après la greffe. Si votre enfant a subi une greffe de rein, votre médecin peut vous demander de prendre ce traitement pendant 200 jours.
Pour les enfants qui ne sont pas en mesure d’avaler le comprimé pelliculé, la poudre pour solution orale peut être utilisée.
Si vous avez pris plus de VALGANCICLOVIR ARROW 450 mg, comprimé pelliculé que vous n’auriez dû :
Contactez immédiatement votre médecin ou un hôpital si vous avez pris, ou pensez avoir pris, plus de comprimés que vous n’auriez dû. La prise d’un nombre trop élevé de comprimés peut provoquer des effets indésirables graves affectant particulièrement votre sang ou vos reins. Une hospitalisation peut être nécessaire.
Si vous oubliez de prendre VALGANCICLOVIR ARROW 450 mg, comprimé pelliculé :
Si vous oubliez de prendre vos comprimés, prenez la dose oubliée dès que vous vous en rappelez et prenez la dose suivante à l’heure habituelle.
Ne prenez pas de dose double pour compenser la dose que vous avez oublié de prendre.
Si vous arrêtez de prendre VALGANCICLOVIR ARROW 450 mg, comprimé pelliculé :
Vous ne devez pas interrompre votre traitement tant que votre médecin ne vous l’a pas demandé.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin ou à votre pharmacien.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Réactions allergiques
Jusqu’à une personne sur 1000 peut avoir une réaction allergique brutale et sévère au valganciclovir (choc anaphylactique). ARRETEZ de prendre VALGANCICLOVIR ARROW et rendez-vous au service des urgences de l’hôpital le plus proche si vous ressentez l’un des troubles suivants :
· éruption cutanée avec démangeaisons (urticaire)
· gonflement brutal de la gorge, du visage, des lèvres et de la bouche pouvant causer une difficulté à avaler ou à respirer
· gonflement brutal des mains, des pieds ou des chevilles.
Effets indésirables graves
· Informez immédiatement votre médecin si vous remarquez l’un des effets indésirables graves suivants - votre médecin pourra arrêter votre traitement par VALGANCICLOVIR ARROW et vous pourrez avoir besoin d’un traitement médical d’urgence :
Très fréquent : peut affecter plus d’une personne sur 10
· faible nombre de globules blancs - avec des signes d’infection tels que des maux de gorge, des ulcères buccaux ou une fièvre ;
· faible nombre de globules rouges - les signes comprennent une sensation d’essoufflement ou de fatigue, des palpitations ou une pâleur de la peau.
Fréquent : peut affecter jusqu’à une personne sur 10
· infection du sang (septicémie) - les signes incluent fièvre, frissons, palpitations, confusion et trouble de l’élocution ;
· faible nombre de plaquettes - les signes comprennent des saignements ou des ecchymoses (« bleus ») plus facilement que d’habitude, du sang dans l’urine ou les selles ou un saignement des gencives ; le saignement peut être sévère ;
· diminution sévère du nombre de cellules sanguines ;
· pancréatite - les signes sont des maux de ventre intenses qui se propagent dans le dos ;
· convulsions.
Peu fréquent : peut affecter jusqu’à une personne sur 100
· Incapacité de la moelle osseuse à produire des cellules sanguines ;
· Hallucinations - entendre ou voir des choses qui n’existent pas ;
· Pensées ou sentiments anormaux, perte de contact avec la réalité ;
· Altération de la fonction rénale.
Les effets indésirables observés lors d’un traitement par le valganciclovir ou le ganciclovir sont décrits ci-dessous.
Autres effets indésirables
· Si vous ressentez l’un des effets indésirables suivants, dites-le immédiatement à votre médecin ou à votre infirmier/ère :
Très fréquent : peut affecter plus d’une personne sur 10
· muguet et candidose buccale ;
· Infection des voies respiratoires supérieures (par exemple : sinusite, angine) ;
· perte d’appétit ;
· maux de tête ;
· toux ;
· sensation d’essoufflement ;
· diarrhée ;
· nausées ou vomissements ;
· douleurs abdominales ;
· eczéma ;
· sensation de fatigue ;
· fièvre.
Fréquent : peut affecter jusqu’à une personne sur 10
· grippe ;
· infection urinaire - les signes comportent de la fièvre, des mictions (action d’uriner) plus fréquentes et des douleurs en urinant ;
· infection de la peau et des tissus sous cutanés ;
· réaction allergique légère – les signes peuvent être des rougeurs, des démangeaisons ;
· perte de poids ;
· idées dépressives, anxiété ou confusion ;
· troubles du sommeil ;
· sensation de faiblesse ou d’engourdissement des mains ou des pieds, ce qui peut affecter l’équilibre ;
· modifications du sens du toucher, fourmillements, picotements, sensation de piqûre ou de brûlure ;
· modifications du goût ;
· frissons ;
· inflammation de l’œil (conjonctivite), douleurs oculaires ou troubles de la vision ;
· douleur dans les oreilles ;
· pression artérielle basse, ce qui peut entraîner des étourdissements ou un évanouissement ;
· difficultés à avaler ;
· constipation, flatulences, indigestion, douleur dans l’estomac, gonflement de l’abdomen ;
· ulcères dans la bouche ;
· résultats de bilans biologiques du foie et du rein anormaux ;
· sueurs nocturnes ;
· démangeaisons, éruption cutanée ;
· perte de cheveux ;
· douleurs dans le dos, douleurs musculaires ou des articulations, spasmes musculaires ;
· sensation d’étourdissements, faiblesse ou malaise général.
Peu fréquent : peut affecter jusqu’à 1 personne sur 100
· sensation d’être agité ;
· tremblements et secouements ;
· surdité ;
· battements cardiaques irréguliers ;
· urticaire, peau sèche ;
· sang dans l’urine ;
· infertilité chez les hommes - voir rubrique « Fertilité » ;
· douleur thoracique.
Un décollement de la rétine (séparation des deux feuillets de la rétine) est apparu uniquement chez les patients atteints du SIDA traités par VALGANCICLOVIR ARROW pour une infection à CMV.
Effets indésirables supplémentaires chez les enfants et les adolescents
Les effets indésirables décrits chez les enfants et les adolescents sont similaires à ceux rapportés pour les patients adultes.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER VALGANCICLOVIR ARROW 450 mg, comprimé pelliculé ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur l’étiquette et sur l’emballage après EXP. La date de péremption fait référence au dernier jour de ce mois.
Ce médicament ne nécessite pas de précautions particulières de conservation.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient VALGANCICLOVIR ARROW 450 mg, comprimé pelliculé
· La substance active est :
Valganciclovir (sous forme de chlorhydrate de valganciclovir).......................................... 450 mg
Pour un comprimé pelliculé.
· Les autres composants sont :
Corps du comprimé : cellulose microcristalline (grade 101 et grade 102), crospovidone (Type B), povidone (K30), stéarate de magnésium.
Pelliculage : hypromellose (3cP, 6cP), dioxyde de titane (E171), le macrogol 400, polysorbate 80, oxyde de fer rouge (E172).
Qu’est-ce que VALGANCICLOVIR ARROW 450 mg, comprimé pelliculé et contenu de l’emballage extérieur
Les comprimés de VALGANCICLOVIR ARROW sont disponibles conditionnés en blisters (Polyamide-aluminium-PVC/Aluminium) et en flacons (PEHD) avec un bouchon en polypropylène.
Boitages :
Blisters : 10, 30, 60 et 100 comprimés pelliculés
Flacons : 60 et 1000 comprimés pelliculés
Titulaire de l’autorisation de mise sur le marché
26 AVENUE TONY GARNIER
69007 LYON
Exploitant de l’autorisation de mise sur le marché
ARROW GENERIQUES
26 AVENUE TONY GARNIER
69007 LYON
APL SWIFT SERVICES (MALTA) LIMITED
HF26, HAL FAR INDUSTRIAL ESTATE, HAL FAR,
BIRZEBBUGIA, BBG 3000.
MALTE
OU
ARROW GENERIQUES
26 AVENUE TONY GARNIER
69007 LYON
OU
GENERIS FARMACEUTICA, S.A.
RUA JOAO DE DEUS, 19,
2700-487 AMADORA,
PORTUGAL
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Ce médicament est autorisé dans les Etats membres de l'Espace Economique Européen sous les noms suivants : Conformément à la réglementation en vigueur.
[À compléter ultérieurement par le titulaire]
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
< {MM/AAAA}>< {mois AAAA}.>
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).